Arene Chromium Complexes: Heterocyclic Chiral Auxiliaries and Synthetic
Targets
E. Peter Kündig,
Gérald Bernardinelli, Dov Beruben, Benoît Crousse, Angelika
Fretzen, Hassen Ratni, Barbara Schnell, and Long-He Xu.
Département
de Chimie Organique, Université de Genève, 30 quai Ernest
Ansermet, CH-1211 Geneva, Switzerland.
Coordination of an arene to the electrophilic Cr(CO)3
group profoundly modifies the arene reactivity. Temporary complexation
thus offers new possibilities of arene functionalization and transformation.
Since the faces of the arene in the complex are no longer equivalent, reactions
occur either on the face opposite to the metal (exo) or on the same
face as the metal (endo). Excellent control of stereochemistry is
often observed in these reactions which further enhances the potential
of this methodology.1 In this article we show
that heterocycles, either as auxiliaries or as synthetic targets, play
a key role in the chemistry of chromium complexed arenes. The subjects
covered are:
1. Phenyl oxazoline complexes are representative examples
of the use of heterocyclic auxiliaries in this area of chemistry.
Topics presented are:
-
The use of oxazoline auxiliaries to direct nucleophilic additions to an
arene activated by the Cr(CO)3 group.
-
Structural insight into the role of the auxiliary and the nature of the
product of nucleophilic addition.
-
Transformation of the nucleophilic addition product into regio- and stereoselectively
substituted alicyclic molecules or into planar chiral complexes: a new
route to o-substituted benzaldehyde complexes.
-
Asymmetric variants of this methodology.
Topics presented are:
-
Diastereospecific inter- and intramolecular aza Diels-Alder reactions
-
First results of Pd-catalyzed intramolecular Heck reactions of planar chiral
arene complexes.
1. Phenyl oxazoline complexes.
1.1. Synthesis of phenyloxazoline Cr(CO)3
complexes, nucleophilic addition, and structural characteristics of a nucleophile
addition product
The tricarbonylchromium complexes 2 of the phenyl oxazolines 1
are orange, air-stable crystalline solids and are readily accessible in
high yields (Scheme 1).2 A wide range of nucleophiles
(alkyl-, vinyl-, aryl lithium reagents, nitrile stabilized carbanions,
a-phenylsulfinyl stabilized carbanions, thioanisole
stabilized carbanions, and ester enolates) react with complex 2a by
addition to the aromatic ring. ortho-Regioselectivity predominates
and is the exclusive mode of addition for small nucleophiles. para-Regioselectivity
can become important with bulky nucleophiles. The ortho-regioselectivity
is thought to be the result of a combination of the electron-withdrawing
effect of the arene substituent and the ability of the nitrogen lone pair
to coordinate the incoming organolithium reagent.
 Scheme 1
Scheme 1
The addition of carbanions to the arene exo-face leads to anionic
(h5-cyclohexadienyl)
Cr(CO)3 complexes which can be isolated
as air sensitive yellow crystals. In applications in synthesis, isolation
is not carried out and the cyclohexadienyl complexes are generated and
reacted in situ with electrophiles. Complex 3 poses an interesting
structural question and its isolation was therefore undertaken. The interest
centers on the fact that complexes 2a-c contain both a p-bound
(Cr(CO)3) and a s-bound
(oxazoline) electrophilic group. Depending on which functional group dominates
reactivity, the structure of 3 could be a 'normal' cyclohexadienyl
Cr(CO)3 complex with the lithium cation
coordinated to a CO ligand oxygen or an aza-enolate with a N-bound lithium
in a N,O-ketene-acetal structure. The former (A) has literature
precedence in the x-ray structure of the addition product of lithium dithiane
to benzene Cr(CO)3,3
whereas the latter structure type (B) has been proposed in addition
reactions of RLi reagents to naphthyl oxazolines.4
In B, the Cr(CO)3 group may be bound
to both the endocyclic diene system and the exocyclic double bond; and
we note that triene Cr(CO)3 complexes with
these bonding characteristics are known.5

Figure 1 shows the crystal structure of product 3a, obtained
by naphthyllithium addition to complex 2a and crystallization in
dioxane. The surprising answer to the above question is that in 3a
both structural types are represented though A clearly dominates.
The lithium ligands are: a carbonyl ligand oxygen, two molecules of solvent
and a water molecule. The water apparently is instrumental as it provides
the link between the predominant cyclohexadienyl structure and the aza-enolate
structure.6 The distance of 2.776(8) Å
between the water oxygen and the oxazoline nitrogen of a second cyclohexadienyl
chromium complex clearly points to O-H.....N=C
hydrogen bridge. Further evidence for the contribution of the aza-enolate
structure is the shortening of the C-C bond between the cyclohexadienyl
and the oxazoline (1.42(1) Å in 3a, 1.470(6) Å in 2a)
and the lengthening of the C=N bond in 3a compared to 2a
(1.29(1) Å vs. 1.260(5) Å).
Figure 1: Crystal structure of complex 3a featuring elements
of both structures A and B (see text) via an intermolecular
CO....Li....OH2....N(oxazoline)
bridge.
Nucleophilic addition to the ortho and to the meta position
of mono-substituted arene Cr(CO)3 complexes
yields planar chiral addition products 3. The two ortho and
the two meta-positions become diastereotopic when the arene carries
a chiral substituent. ortho-Directing chiral substituents have been
applied with much success (90-100% de) in this reaction.7
We also note the extension of this methodology to diastereoselective meta-addition
reactions.8 Not surprisingly, in meta
additions the levels of induction are considerably lower than in ortho-additions
(48% de 8a ; up to 76% de 8b,c
).
Chiral hydrazones and chiral oxazolines have been shown to be excellent
diastereoselective o-directing groups. With L-valinol and L.-tert-butylglycinol
derived oxazolines, the product stereochemistry (see 1.2.2-1.2.4)
reflects a transition state in which the substituent at the stereogenic
center of the auxiliary is syn to the metal fragment. In the anti-rotamer,
the lithium coordination controlled addition of the C-nucleophile would
lead to a more congested transition state as shown below. The X-ray structure
of complex 2b (R = i-Pr) shows both an endo- and an
exo-rotamer in the unit cell. It also shows that the i-propyl
group in the endo-rotamer is far from the Cr(CO)3
group. It is therefore unlikely that one of the rotamers is largely favored
in solution. The structure further reveals that the pseudo-equatorial i-propyl
group may not be bulky enough to completely prevent nucleophilic addition
to the arene in the exo-rotamer (hence diastereomeric excesses around
90%). Indeed, its replacement by a t-Bu group (complex 2c
(R1 = t-Bu, R2 = H))
leads to considerable improvement of diastereoselectivity
(see 1.2.3.).
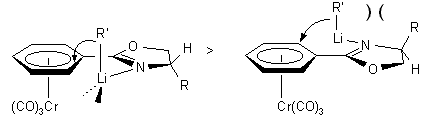
Figure 2: Crystal structure of complex 2b
In a complementary approach, regio- and enantioselective addition of
alkyl-, vinyl- and aryllithium reagents to prochiral (h6-arene)Cr(CO)3
complexes was achieved in the presence of an external chiral ligand. The
best results were obtained with the readily accessible C2-chiral
dimethoxydiphenylethane. Some examples of these reactions with complex
2a will be shown in sections 1.2.1 and 1.2.4. It should be pointed
out that this methodology has the potential to be catalytic in the added
chiral ligand.9
1.2. Reactions of complexes 3 with electrophiles
The anionic cyclohexadienyl complex 3a reacts with electrophiles
to give complexed or decomplexed substituted arenes or cyclohexadienes.
An overview is shown in Scheme 2.
 Scheme 2
Scheme 2
Examples of these selective transformations are given in Schemes 3-10.
1.2.1. Nucleophile addition/oxidation reactions with complex 2a
Formally, this sequence is a nucleophilic substitution of a hydride by
a carbanion. Its merits are a) a regioselectivity that is often complementary
to electrophilic substitution and b) a reaction that takes place under
very mild conditions yielding substituted aromatics in high yield.1k
Although the mechanism has not been established, a reasonable sequence
involves metal oxidation in complex 3 followed by intramolecular
C(6)-H(endo) hydride transfer to the metal and oxidative cleavage
of the metal arene bond. Applied to complex 2a a range of o-disubstituted
arenes 4 have been obtained.10
 aAfter equilibration of the intermediate. bo/p
= ca. 4:1.
Scheme 3
aAfter equilibration of the intermediate. bo/p
= ca. 4:1.
Scheme 3
1.2.2. Nucleophile addition/hydride-abstraction reactions
with complex 2a
A recent reinvestigation of the feasibility of endo-hydride abstraction
in anionic cyclohexadienyl chromium complexes resulted in a reaction protocol
for the transformation 2 ® 5 shown
in Scheme 4. The highly regioselective nucleophilic aromatic substitution
of a hydride for a carbanion6 in benzaldehyde
imine, hydrazone and, as shown here, in phenyl oxazoline complexes, yields
planar chiral arene complexes. It shows that the metal arene bond can indeed
be conserved in the aromatic substitution and used in subsequent metal
activated transformations.
 Scheme 4
Scheme 4
An enantioselective synthesis of planar chiral complexes is based on
the chiral external ligand mediated nucleophile addition mentioned in section
1.1 (Scheme 5).11
 Scheme 5
Scheme 5
1.2.3. Nucleophile addition/allylation or propargylation
with complexes 2a - 2c.
In situ reaction of the anionic cyclohexadienyl complexes 3 with
allyl or propargyl bromides yields dienes 6.12
The trans stereochemistry between nucleophile and allyl- or propargyl
groups was established by 1H-NMR and confirmed by an X-ray structure
determination. It suggests a sequence of allylation/propargylation at the
metal followed by reductive elimination and decomplexation.
 Scheme 6
Scheme 6
Yields with propargyl bromides are usually higher (>70%) than with allyl
and benzyl bromides (<70%) since with the latter, the crude products
also contain 10-20% of ketone products resulting from migratory CO insertion
prior to reductive elimination.
Figure 3: Crystal structure of cyclohexadiene 6a resulting from
the addition of PhLi and allyl bromide to the phenyl oxazoline Cr(CO)3
complex 2a.
Cyclohexadienes 6 offer a number of possibilities for polar,
radical13 and transition metal mediated14-15
intramolecular cyclization reactions.

As an example we show in Scheme 7 the transformation of the alkynyl
moiety in 6b to a vinylic radical which then undergoes a 5-exo-trig
cyclization. Depending on R in 6b the reaction goes through one
or the other of the two regioisomeric vinylic radical intermediates and
this then determines the cyclization to either C(1) or C(4).16
 Scheme 7
Scheme 7
Examples of diastereoselective syntheses of enantioenriched cyclohexadienes
are shown in Scheme 8 7 and Scheme 9.
 Scheme 8
Scheme 8
 Scheme 9
Scheme 9
1.2.4. Nucleophile addition/acylation/alkylation
In contrast to propargyl- and allyl halides, alkyl bromides and iodides
react with 3 to yield ketone products. Metal alkylation is thus
followed by migratory carbonylation prior to reductive elimination. An
added alkylation step completes the one-pot reaction which consists of
forming in a highly regio-and diastereoselective way three new C-C bonds
and two new stereogenic centers (Scheme 10, Figure 4).12
Again, diastereoselective non-racemic versions have been demonstrated.
 Scheme 10
Scheme 10
Figure 4: Crystal structure of cyclohexadiene 7a (see Scheme
10).
Conversion of the oxazoline function in complex 5 by alkylation/reduction
or carrying out the same chemistry described above with benzaldehyde imine
or hydrazone complexes yields, after hydrolysis, highly enantioenriched
benzaldehyde complexes. The nucleophile addition, hydride abstraction sequence
in Scheme 5 provides a new enantioselective entry to these compounds.17
We take the opportunity of this symposium to report on two applications
of these complexes in aza-heterocycle synthesis.
2. Cycloaddition and cyclization reactions involving planar
chiral benzaldehyde imine complexes.
2.1. Diastereoselective intra- and intermolecular aza-Diels-Alder reactions
2.1.1. Intramolecular cycloaddition. Chiral aryl-2-aza-diene complex.
The enantiopure complex 8 was readily obtained from the corresponding
aldehyde complex using a literature method.18
Lewis acid mediated cycloaddition and decomplexation afforded a 4: 1 mixture
of the enantiopure diastereoisomers 9 and 10 (Scheme 11).
This result shows that the facial selectivity of the diene is perfectly
controlled by the chiral complex whereas the exo/endo selectivity
of the dienophile is not as good as hoped for.19
 Scheme 11
Scheme 11
2.1.2. Intermolecular cycloaddition: chiral benzaldehyde imine complex
as dienophile
The planar chiral benzaldehyde imine complexes undergo Lewis acid mediated
aza-Diels-Alder reactions with Danishefsky's diene to give the dihydropyridinones
with fair to excellent diastereoselectivity.18,20
(Scheme 12). Radical cyclization of the enantiopure complexes 11e
and 11f yield quinolizidines 12 (Scheme 13) and indolizidines
(not shown).18
 Scheme 12
Scheme 12
 Scheme 13
Scheme 13
2.2. Pd-catalyzed intramolecular Heck reactions of planar chiral arene
complexes
We have recently reported the first intramolecular Heck-reactions with
planar chiral arene Cr(CO)3 complexes21
and have now extended these studies to the dihydropyridinone complexes
11c,d. By carefully controlling the reaction conditions these cyclizations
can be carried out without decomplexation from the Cr(CO)3
unit and without alkene isomerization. (Scheme 14).
 Scheme 14
Scheme 14
Acknowledgements
We thank the Swiss National Science Foundation for financial support of
this work.
We gratefully acknowledge the help of Carmine Chiancone in the construction
of these Web pages.
References
-
a) W. E. Silverthorn, Adv. Organomet. Chem. 1975,
13, 48; b) G. Jaouen in Transition Metal Organometallics in Organic
Synthesis, Vol. 2, (Ed.: H. Alper), Academic Press: New York (USA),
1978, p. 65; c) S. G. Davies in Organotransition Metal Chemistry,
Applications in Organic Synthesis, Pergamon Press:
Oxford (UK), 1982, p. 166; d) A. Solladie-Cavallo, Polyhedron
1975, 4, 901; e) E. P. Kündig, Pure Appl. Chem.
1985, 57, 1855; f) V. N. Kalinin, Russ. Chem. Rev.
1987, 56, 682; g) L. Balas, D. Jhurry, L. Latxaoue, S. Grelier,
Y. Morel, M. Handani, N. Ardoin, D. Astruc, Bull. Soc. Chim. Fr.
1990, 127, 401; h) P. J. Dickens, J. P. Gilday, J. T. Negri,
D. A. Widdowson, Pure Appl. Chem. 1990, 62, 575; i)
J. McQuillin, D. G. N. Parker, G. R. Stephenson in Transition Metals
in Organic Synthesis, 1st ed., Cambridge University Press: New York
(USA), 1991, p. 182; j) S. L. Hegedus, Transition Metals in the
Synthesis of Complex Organic Molecules, University Science Books: Mill
Valley (California/USA), 1994; k) M. F. Semmelhack
in Comprehensive Organometallic Chemistry II (Eds.: E. W. Abel,
F. G. A. Stone, G. Wilkinson), Vol. 12 (Ed.: L. S. Hegedus), Pergamon Press:
Oxford (UK), 1995, p. 979; l) ibid., p. 1017; m) M. J. Morris
in Comprehensive Organometallic Chemistry II (Eds.: E. W. Abel,
F. G. A. Stone, G. Wilkinson), Vol. 5 (Eds.: J. A. Labinger, M. J. Winter),
Pergamon Press: Oxford (UK), 1995, p. 393; n) S. G. Davies, T. D.
McCarthy in Comprehensive Organometallic Chemistry II (Eds.: E.
W. Abel, F. G. A. Stone, G. Wilkinson), Vol. 12 (Ed.: L. S. Hegedus), Pergamon
Press: Oxford (UK), 1995, p. 1039; o) M. Uemura, Rev. Heteroatom
Chem. 1994, 10, 251.
-
E. P. Kündig, A. Ripa, R. Liu, D. Amurrio, G. Bernardinelli,
Organometallics 1993, 12, 3724.
-
M. Semmelhack, H. T. Hall, R. Farina, M. Yoshifuji, G.
Clark, T. Bagar, K. Hirotsu, J. Clardy, J. Am. Chem. Soc. 1979,
101, 3535.
-
T. G. Gant, A. I. Meyers, Tetrahedron 1994,
50, 2297.
-
J. Blagg, S. G. Davies, C. L. Goodfellow, K. H. Sutton,
J. Chem. Soc., Perkin Trans. I 1990, 1, 1133 and ref.
cited.
-
A. Fretzen, A. Ripa, R. Liu, G. Bernardinelli, E. P. Kündig,
Chem. Eur. J. 1998, 4, 251.
-
E. P. Kündig, A. Ripa, G. Bernardinelli, Angew.
Chem. Int. Ed. 1992, 31, 1071.
-
a) M. F. Semmelhack, H.-G. Schmalz, Tetrahedron Lett.
1996, 37, 3089; b) A. J. Pearson, A. V. Gontcharov, Tetrahedron
Lett. 1996, 37, 3089; c) A. J. Pearson, A. V. Gontcharov,
J. Org. Chem. 1998, 63, 152.
-
For external chiral ligand controlled nucleophilic additions
see: a) H. Fujieda, M. Kanai, T. Kambara, A. Iida, K. Tomioka, J. Am.
Chem. Soc. 1997, 119, 2060 and ref. cited; b) S. E. Denmark,
N. Nakajima, O. J.-C. Nicaise, J. Am. Chem. Soc. 1994, 116,
8797; Review in: S. E. Denmark, O. J.-C. Nicaise, J. Chem. Soc., Chem.
Commun. 1996, 999.
-
E. P. Kündig, A. Ripa, R. Liu, D. Amurrio, G. Bernardinelli,
Organometallics 1993, 12, 3724.
-
A. Fretzen, E. P. Kündig, Helv. Chim. Acta
1997, 80, 2023.
-
E. P. Kündig, A. Ripa, R. Liu, G. Bernardinelli,
J. Org. Chem. 1994, 59, 4773.
-
For a review see: B. Giese, B. Kopping, T. Göbel,
J. Dickhaut, G. Thoma, K. J. Kulicke and F. Trach, Org. React. 1996,
48, 301.
-
a) L. S. Hegedus in Transition Metals in Organic Chemistry,
University Science Books: Mill Valley (California/USA), 1994; b)
J. Tsuji in Palladium Reagents and Catalysts, John Wiley & Sons:
Chichester (UK), 1995.
-
A. de Meijere, F. E. Meyer, Angew. Chem. Int. Ed.
1994, 33, 2379.
-
D. Beruben, E. P. Kündig, Helv. Chim. Acta.
1996, 79, 1533.
-
Other available methods: Resolution: a) A. Solladié-Cavallo,
G. Solladié, E. Tsamo, J. Org. Chem. 1979, 44,
4189; idem, Inorg. Synth. 1985, 23, 85; b) L. A. Bromley,
S. G. Davies, C. L. Goodfellow, Tetrahedron Asymmetry 1991,
2, 139; c) C. Baldoli, P. Del Buttero, S. Maiorana, Tetrahedron
1990, 46, 7823; diastereoselective complexation of arenes
bearing chiral auxiliaries: d) A. Alexakis, P. Mangeney, I. Marek, F. Rose-Munch,
E. Rose, A. Semra, F. Robert, J. Am. Chem. Soc. 1992, 114,
8288; diastereoselective ortho-lithiation: e) J. A. Heppert, J.
Aubé, M. E. Thomas-Miller, M. L. Milligan, F. Takusagawa, Organometallics
1990, 9, 727; f) J. Aubé, J. A. Heppert, M. L. Milligan,
M. J. Smith, P. Zenk, J. Org. Chem. 1992, 57, 3563;
g) Y. Kondo, J. R. Green, J. Ho, J. Org. Chem. 1993, 58,
6182; h) P. W. N. Christian, R. Gil, K. Muñiz-Fernández,
S. E. Thomas, A. T. Wierzchleyski, J. Chem. Soc., Chem. Commun.
1994, 1569; i) A. Alexakis, T. Kanger, P. Mangeney, F. Rose-Munch,
A. Perrotey, E. Rose, Tetrahedron Asymmetry 1995, 6,
47; ibid 2135; j) J. W. Han, S. U. Son, Y. K. Chung, J. Org.
Chem. 1997, 62, 8264; enantioselective lithiation: k)
E. P. Kündig, A. Quattropani, Tetrahedron Lett. 1994,
35, 3497.
-
E. P. Kündig, L.-H. Xu, P. Romanens, G. Bernardinelli,
Synlett 1996, 3, 270.
-
For a report of a highly diastereoselective intramolecular
2-azadiene cycloaddition reaction with an arene Cr(CO)3
complex see: S. Laschat, R. Noe, M. Riedel, Organometallics 1993,
12, 3738.
-
C. Baldoli, P. Del Buttero, M. Di Ciolo, S. Maiorana,
A. Papagni, Synlett 1996, 258.
-
B. Crousse, L.-H. Xu, G. Bernardinelli, E. P. Kündig,
Synlett 1998, 658.
(c) ECHET98. June 1998