Instructions for Experiment 3: Chemical Informatics
These pages represent detailed instructions for the techniques described. Some
techniques are deemed sufficiently "intuitive" that no details are given here.
In other cases, the information provided by the supplier itself is sufficient,
and is not replicated here. To access each individual information point, click on
the icon you see. To find out more about the information provider, click on the blue
hyperlink next to the icon. The strip  means you can return to
an overview of the "Information booth".
means you can return to
an overview of the "Information booth".


 This runs a terminal emulation program called Telnet. Login with
account iic02p (the password is available from members of staff) and select the ISI service.
The menus are largely driven by typing appropriate characters from the
keyboard. In this instance, they are case insensitive.
This runs a terminal emulation program called Telnet. Login with
account iic02p (the password is available from members of staff) and select the ISI service.
The menus are largely driven by typing appropriate characters from the
keyboard. In this instance, they are case insensitive.




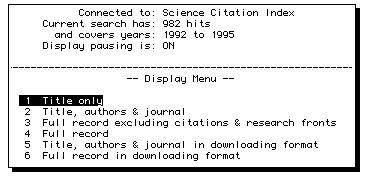
More detailed instructions are summarised in a small manual
available from the
chemistry library or the on-line office in the Lyon Playfair laboratory.

CAS On-line
This will enter a Telnet session. Type z as the first thing you enter, followed by
an account number and password (this is available on request) followed by 3 for the
terminal type.
You will next have to specify the database file you want by type FILE CA (the Main
CAS bibliographic file)
or FILE REG (the Registry file of substances) or FILE LCA (The learning CA file).
Entry to these files is only at certain times of day (normally AM). Only the LCA
file has no associated cost, all other searches will accumulate a charge which will have
to be paid by the owner of the account number.

Silver Platter
This offers "samplers" of a number of
commercial databases. The full versions of several of these are also available
via dedicated software, which on a Macintosh is known as "MacSPIRS".




Libertas
Again the Telnet program is used to access the College catalogues;



 A major chemical supplier has made available their
available chemicals directory.
A major chemical supplier has made available their
available chemicals directory.

 A major chemical supplier has made available their
available chemicals directory. One of the "value added services they offer
is hazard safety sheets for all their entries. You can take advantage of
this by searching for information on any penicillins.
A major chemical supplier has made available their
available chemicals directory. One of the "value added services they offer
is hazard safety sheets for all their entries. You can take advantage of
this by searching for information on any penicillins.

 This information provider was one of the first
companies to offer a "Web" interface to their databases. Here you can see it
in action on "sampler" datasets. A number of interfaces are offered in this
service, including the World Drug Index, and the "Savant" system.
This information provider was one of the first
companies to offer a "Web" interface to their databases. Here you can see it
in action on "sampler" datasets. A number of interfaces are offered in this
service, including the World Drug Index, and the "Savant" system.

Start with the WDI database, entering a suitable search term;

The results of a keyword search are displayed on the screen.
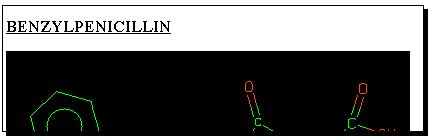
Clicking
on the "thumbnail" graphic of any molecule found will reveal
further properties, one of which is the so-called SMILES
string. This is a powerful and popular method of representing
molecular structure as a sequence of simple characters. It
is also one of the few methods for transferring molecular
structure definitions between different programs. To illustrate
how this works, select the SMILES string shown below the
structure, and "copy" it to the clipboard using the "edit" menu.
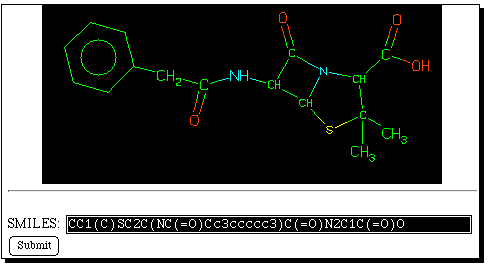
Now select the "back" button on the WWW browser, and enter
instead the "Savant" database. The query SMILES string
can be pasted into the keyword search field. Savant produces
a "similarity" search which finds bibliographic references
to recent literature relating to the synthesis of compounds
either identical to, or similar to the SMILES search query.
 Select a compound which may look interesting, and
investigate one or two recent literature references to
its synthesis;
Select a compound which may look interesting, and
investigate one or two recent literature references to
its synthesis;

 This experiment should ideally be performed on the Silicon
Graphics computers. If you use a Macintosh, a program called MacX
will start up, and following prompts, you will need to enter your SGI
account and its password. On the SGI, type in response to the prompts that
appear;
This experiment should ideally be performed on the Silicon
Graphics computers. If you use a Macintosh, a program called MacX
will start up, and following prompts, you will need to enter your SGI
account and its password. On the SGI, type in response to the prompts that
appear;
>MENU
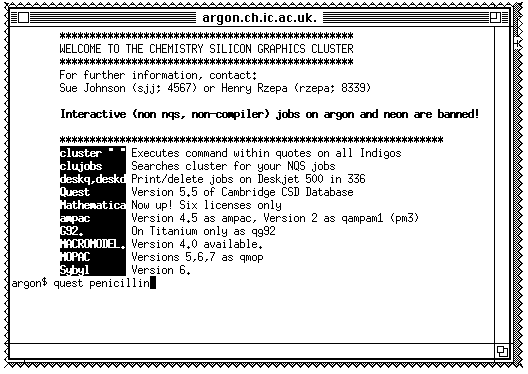
On a Macintosh computer, enter the line
quest penicillin
in
the MacX window that appears, followed by
TERM X11
MENU
You will need to search the Cambridge
crystal structure database for the penicillin and cephalosporin sub-structures.
The first menu allows a
molecule to be drawn. You will notice that there is no common standard for
drawing molecules on screen (unfortunately). With Quest, a click
on the screen draws the atom selected (by default C,
but in the example below
currently S). Further clicks add a further atom to the last atom. If you
want the next atom not to be added to the previous, select MOVE first. To
convert a single to a double bond, select DOUB, then MOVE and click on the bond
desired. To convert a carbon to another atom on the menu, select the type from
the screen menu and then MOVE before clicking at the desired atom. If the atom
is not present on the menu, click on OTHER. Errors can be corrected using
DELATOM or DELBND, or in extremis CLEAR which removes the entire molecule.

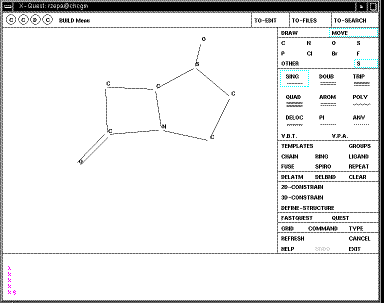
When you are happy with your structure, click on DEFINE STRUCTURE and then
QUEST;
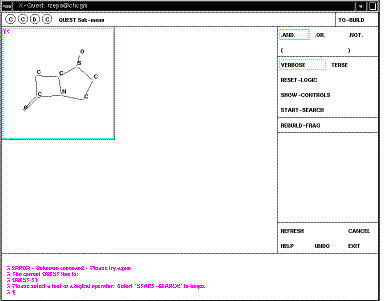
Your defined search operators are shown as T1 (T2 etc). In this case they are
sub-structures, but many other definitions are possible, including author
names, formulae, etc. It is possible to combine several of these using logical
operators. In this case none of this is necessary. Just click on the T1 box to
select this item, then START-SEARCH. As soon as a "hit" is found, it is
displayed on the screen;
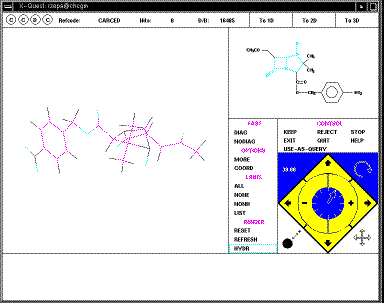
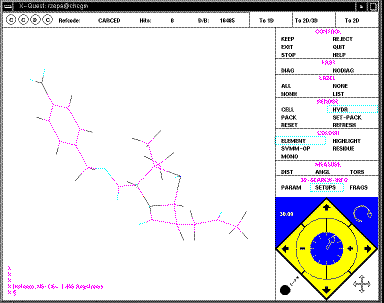
The structure can be rotated using the four small arrows in the bottom right
corner of the menu (you might wish to discuss in your writeup the pros and cons
of this method compared with those found in other programs; again no common
standard applies).
At this point, you have to either KEEP or REJECT the
structure before the next one is displayed. If you KEEP the structure, its
co-ordinates are written to a file on disk (called penicillin.dat in
this case). Continue to the end, when the exit from the database will occur. To
perform a further search, type Quest <name of search> from the console
window of the workstation, as shown in an earlier diagram above. At any stage
you can also go to the alternative 3D projection, in which bond lengths, angles
etc can be displayed on the screen.
 This enables a simple keyword search of the PDB archives, with the result
displayed in a window on your screen. Be aware that the PDB files can be quite
large, and that response times for their retrieval are likely to be better in
the morning than the afternoon. A mor sophisticated interfaces is avalailable
via the European Bioinformatics Institute.
This enables a simple keyword search of the PDB archives, with the result
displayed in a window on your screen. Be aware that the PDB files can be quite
large, and that response times for their retrieval are likely to be better in
the morning than the afternoon. A mor sophisticated interfaces is avalailable
via the European Bioinformatics Institute.






 When Beilstein Commander starts up, you will be confronted by a login-prompt. Use
chsr as the account. The password is available from staff.
When Beilstein Commander starts up, you will be confronted by a login-prompt. Use
chsr as the account. The password is available from staff.
 Due to a programming error in this program, the molecule window is empty.
Click on a second
time, and this will be replaced with a molecule query.
Due to a programming error in this program, the molecule window is empty.
Click on a second
time, and this will be replaced with a molecule query.
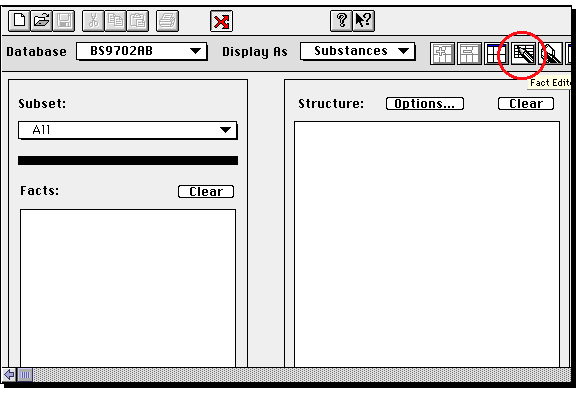
 If you wish to edit or modify this structure, double click on the molecule, whereupon
a structure editor window will open;
If you wish to edit or modify this structure, double click on the molecule, whereupon
a structure editor window will open;
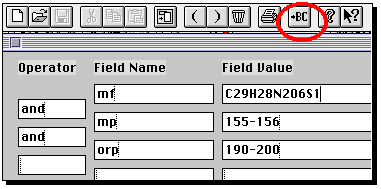
 The important thing about this structure, is that missing valencies are assumed
to be hydrogen rather than generic substituents. To allow a search to proceed
on the assumption that any substituent can occupy a free site, go to the capture tool
(the dotted square icon), then highlight an atom (or several, by dragging the capture
box) and from the Query menu, select "Free Sites". This places a star against the atom(s).
If you do not do this, you may not find any structures. Other editing operations are
(sort of) intuitive, and it is suggested you explore them. Once happy with the structure,
click on the "BC" button along the top to return to Commander.
The important thing about this structure, is that missing valencies are assumed
to be hydrogen rather than generic substituents. To allow a search to proceed
on the assumption that any substituent can occupy a free site, go to the capture tool
(the dotted square icon), then highlight an atom (or several, by dragging the capture
box) and from the Query menu, select "Free Sites". This places a star against the atom(s).
If you do not do this, you may not find any structures. Other editing operations are
(sort of) intuitive, and it is suggested you explore them. Once happy with the structure,
click on the "BC" button along the top to return to Commander.
 Now click on Start to
commence the search.
You will be told how many hits are found (for an unmodified structure
query it should be between 100-200!). A new program called Display Hits starts up
(by this time, your Macintosh might be suffering from a lack of memory, and you
may need to shut down any non-essential programs). From menu View, select Short display to
get a preview of structures found.
Now click on Start to
commence the search.
You will be told how many hits are found (for an unmodified structure
query it should be between 100-200!). A new program called Display Hits starts up
(by this time, your Macintosh might be suffering from a lack of memory, and you
may need to shut down any non-essential programs). From menu View, select Short display to
get a preview of structures found.
 You can select individual entries by clicking on them.
Go back to full display to obtain data such as optical rotation or melting point.
You can select individual entries by clicking on them.
Go back to full display to obtain data such as optical rotation or melting point.

 This section will show how structural information from the Daylight WDI
and Savant database and the Cambridge CCDC searches can
be transferred to a local computer using a simple program
such as Telnet, and used to
generate a local database.
This section will show how structural information from the Daylight WDI
and Savant database and the Cambridge CCDC searches can
be transferred to a local computer using a simple program
such as Telnet, and used to
generate a local database.
Part 1:
There are several ways of transferring a file
from a remote computer to the one you are using. The method that follows is the oldest, but
in some ways the simplest and probably the fastest.
Login to the Unix cluster
using your own account and password, making use of the Telnet program
invoked by clicking on the icon above. Firstly, enable file transfer if it is not already;
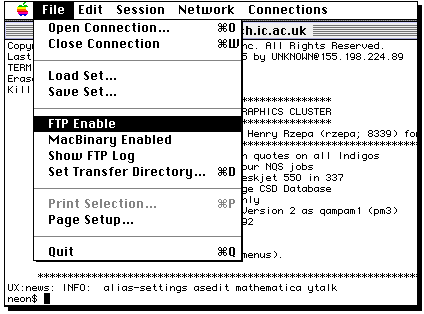
Type ftp, then a space, then select the IP number;
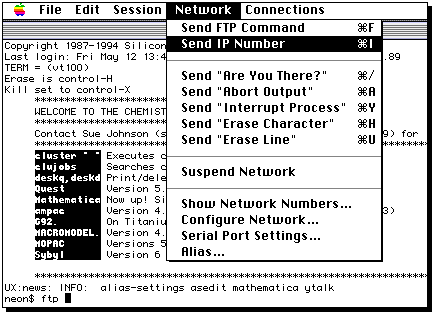
Select the Macintosh directory where the file will go;
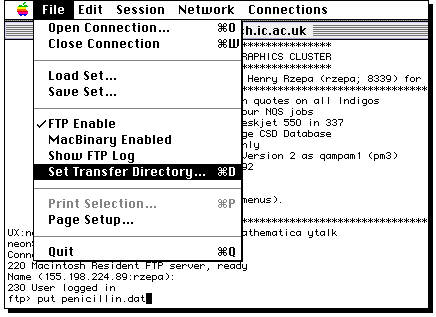
Press the RETURN key, when the file will be transferred.
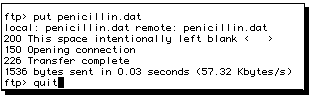
Part 2:
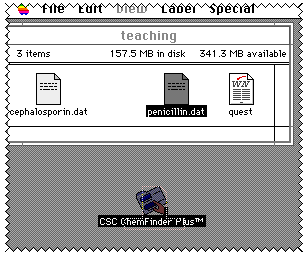
A file called "penicillin.dat" (or whatever name you used) should now
be found in the directory you chose to put it.
Selecting this file and drag it
over an Icon called ChemFinder located on the desktop of the Mac you are using
and release the mouse button.
You will be asked to identify the type of file being processed;
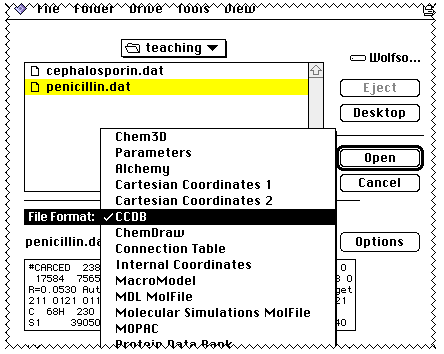
If the conversion is successful, an entry will be created in a database called
ChemFinder;
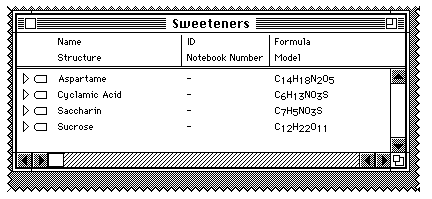
The analogy (or "metaphor") of this database is that each collection of
chemical information is stored in a folder. In your case, when the folder is
"double clicked" you get to see the entries in it, in this case one compound.
Each compound has a triangle on the left, which if clicked points down, and
various attributes of the molecule are revealed. In your case, you should see a
ChemDraw 2D representation and a Chem3D picture, in addition to the formula
already displayed;
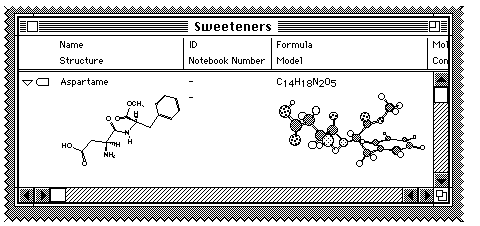
Double clicking in the centre of either "cell" (to use spreadsheet analogy)
will enable you to edit that entry using either ChemDraw or Chem3D (again just
like a spreadsheet). If you double click on the 3D diagram, you can create an
animation of the molecule as shown below;
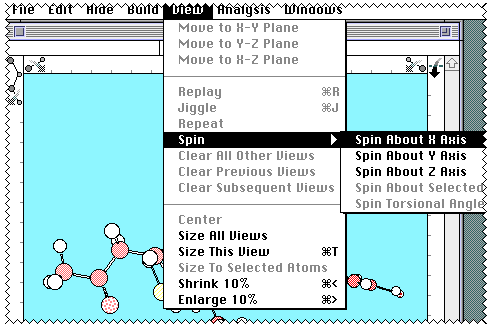
Also whilst in Chem3D, you may wish to select the molecule, copy to
the clipboard, and paste it into your report, or print it
directly on any printer attached to the Macintosh.
 The action of clicking on the ISIS logo should activate a program
called ISIS/Draw. This should already contain a reaction query defined
for you.
The action of clicking on the ISIS logo should activate a program
called ISIS/Draw. This should already contain a reaction query defined
for you.
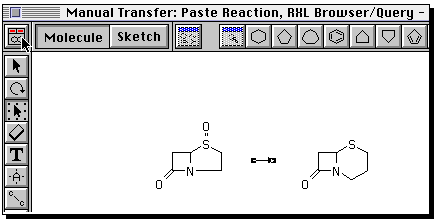
You can if you wish define an entirely new reaction. To do so,
proceed as follows. Build the reactant using ring templates,
and suitable bond tools. To insert heteroatoms, select
the "A" tool, click on the atom, when a small box should appear,
and type the atom symbol from the keyboard.
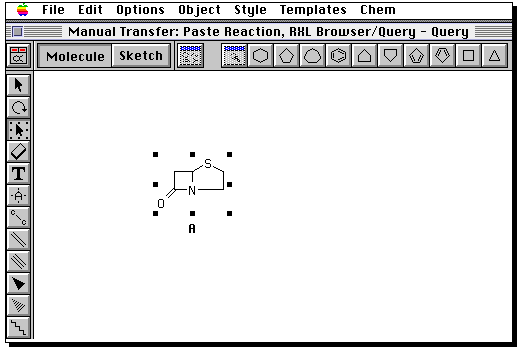
Draw the product in the same manner. Then go to the "box" tool,
and "select all" from the edit menu. Now from the "Chem" menu,
select "reaction".
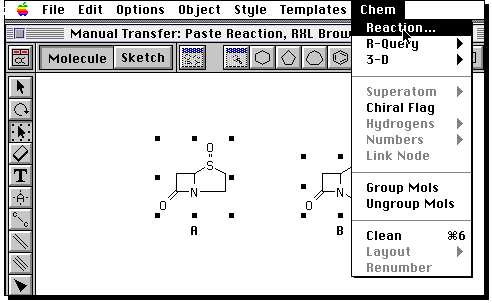
You now have to define the relationship of the two molecules
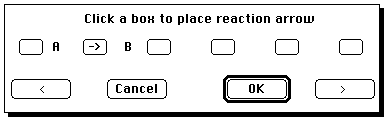
A reaction is defined. Now you have to launch ISIS/Base. Because this
requires a lot of memory, you will have to close any other applications
that might be running (including Netscape!). Launch the program from the
Apple menu in the "chemistry programs" sub-menu. Once open, select
"database" from the "file" menu. Open RXN browser. You will need to
provide a user number and password. Ask a member of staff for this.
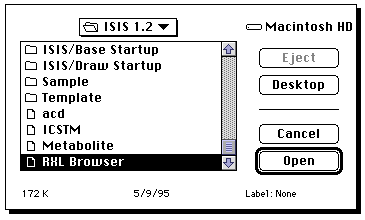
Once Base is up and running (it may take about 2-3 minutes to achieve this
state) return to the Draw program. Select ALL and then COPY from the Edit menu,
return to Base, and PASTE the diagram in.
The reaction should now appear in the Base window;
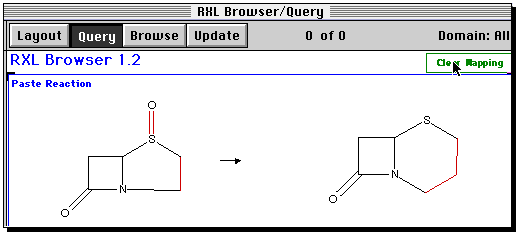
Some bonds will be highlighted to indicate a possible
bond mapping between the two structures. This guess is likely
to be wrong, so the safest course of action is to clear mapping.
Now from the Search menu, select RSS (reaction sub-structure);
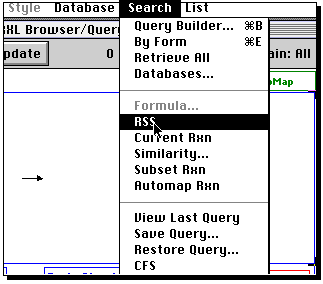
A list of "hits" will appear. Select the "su" button, and if you wish
scroll down the list of hits with the "down" button. If you wish to
copy an entire hit, select the "copy form" item from the edit menu,
and then paste this into a word processor. (since you may have stopped
all other programs due to memory, you may now have to start Claris up again).
 This represents a virtual library of structural and chemical informatio, presented
in the style of a textbook of "knowledge". It represents the final stage of
the transition from data to knowledge. Identify if any contents are relevant
to penicillin.
This represents a virtual library of structural and chemical informatio, presented
in the style of a textbook of "knowledge". It represents the final stage of
the transition from data to knowledge. Identify if any contents are relevant
to penicillin.

Electronic Conferences
Unlike real conferences, electronic versions can be easily
indexed. The ECTOC conference, hosted by Imperial College during
June-July of 1995, was the first such conference to support
index searching. Find out if penicillin was mentioned!
 Browse through the Chemical Copmmunications
or
Network Science to get a typical impression of
these new media.
Browse through the Chemical Copmmunications
or
Network Science to get a typical impression of
these new media.
