Crown ethers are well recognised as reversable binding sites for a wide variety of cations, from simple metal ions such as Na+ and K+ to more complex quaternary ammonium ions.[1] Supermolecular structures containing two or more crown ether functionalities have attracted wide interest, particularly in the field of artificial receptor synthesis.[2-10]Certain of these compounds have shown considerable promise as trace metal ion detectors[5] while others display selective cation binding dependant upon the presence of other bound ions.[2,10] The majority of known bis (crown ether) receptors possess a flexible link between the functionalities or are incorporated on either side of a calixarene.[8] We report here the selective synthesis of a bis (crown ether) where the functionalities are rigidly held on an alicyclic framework.
Ring-strained alkenes are reactive cycloaddition reagents known to react with regular or inverse electron-demand dienes.[11] Cyclic diazadienophiles such as phthalazinediones, on the other hand, are extremely potent dienophiles yet react only with regular electron-demand dienes.[12] We reasoned that these two features could be exploited by using the phthalazinediones as carriers of functionality (effectors) and converting them to ring-strained alkenes prior to their use as BLOCK reagents in our regular electron-demand ACE (alkene plus cyclobutene epoxide)[13,14] coupling protocol. In this way, the phthalazinedione can be trapped and stored as alkene BLOCKs, suitable for attachment to the full range of 1,3-diene or 1,3-dipolar BLOCK partners. We demonstrate this concept by using phthalazinediones to carry the crown ether functionality in the construction of new bis (crown ethers).[13,14]
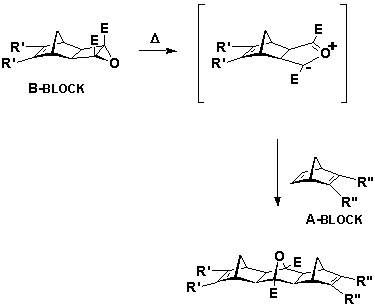
There are two different types of mono BLOCKs applicable for the ACE reaction.[13,14] Type A-BLOCKs which contain an olefin moiety, typically a substituted norbornene group and type B-BLOCKs which contain a cyclobutene epoxide functionality. The advantage of the ACE reaction is found in its selectivity, with both types of BLOCKs necessary for a successful reaction, and neither A nor B-BLOCKs able to react in the absence of the other, Scheme 1. Futhermore, adducts of the ACE reaction, which may be conducted under either thermal or photochemical conditions,[13,14] show primarily exo,exo stereochemistry.[15]
Scheme 1 E=CO2Me
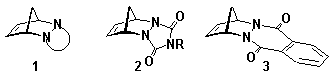
A key requirement of the BLOCK coupling reactions is knowledge of the stereochemistry of the individual BLOCKs, especially the positioning of the functionality (effector) carried by the BLOCK. The stereochemistry of nitrogen atoms in ring-fused derivatives of cyclic diazadienophiles, eg cyclopentadiene adducts of type 1, are known from X-ray studies[16] to differ depending on the size of the original heterocyclic ring. The ring-junction nitrogens exhibit pyrimidal stereochemistry in triazoline dione adducts of type 2 (5-membered ring)[16] whereas the nitrogens in related phthalazinedione adduct 3 (6-membered ring) display near-planar stereochemistry.[16] This feature has been exploited in the chemistry described in the present article, where the activity of the alkene p-bond in 3 and its derivatives in ACE coupling reactions has provided new ribbon systems in which the planar stereochemistry[16] of the ring-nitrogens defines the stereochemistry of the benzenoid portion of the adduct.
Scheme 2
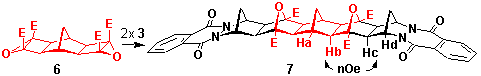
Model studies using the parent phthalazinedione adduct 3 established the ability of diazanorbornenes to participate in the ACE coupling reaction, Scheme 3. Heating 3 with the cyclobutene epoxide 4 containing the dimethoxynaphthalene chromophore(sealed tube, dichloromethane, 140oC) yielded the coupled product 5 in 55% yield. The yield was improved to 80% by conducting the reaction under photochemical conditions (acetone, 300 nm, Rayonet reactor), Scheme 3.
That dual ACE coupling could also be achieved was established by reaction of the phthalazinedione adduct 3 with the bis(cyclobutene epoxide) 6 under thermal conditions (dichloromethane, 140oC, 4h, 64%) (Scheme 3). The resultant product 7 exhibited C2v symmetry typified by the presence of only four methine resonances Ha=d2.17 (s, 2H), Hb=d2.72 (s, 4H), Hc=1.95 (s, 4H), Hd=5.11 (s, 4H), a single ester resonance at d=3.94 (12H), bridge methylenes at d1.92 (s, 2H) and d1.78, 2.87 (dd J=10.8 Hz, 4H), and aromatic resonances at d7.78, 8.26 (m, 8H), while the exo, exo-stereochemistry was confirmed by an nOe between Hb and Hc.

Scheme 3 E=CO2Me
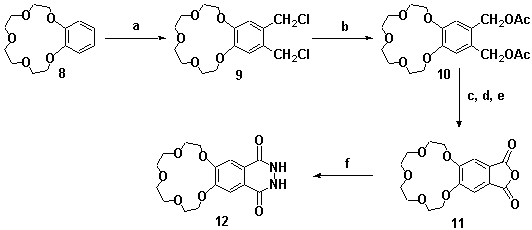
To incorporate a crown ether functionality into ribbon molecules of type 5 and 7, the 15-crown-5 analogue 14 of A-BLOCK 3 was synthesised. Starting from benzo-15-crown-5 8, phthalhydrazide 12 was synthesised in four steps via methodology adapted from Lehn and co-workers,[7] Scheme 4.
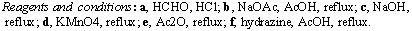
Scheme 4
Synthesis of the diazanorbornene containing crown ether 14 was achieved via the in situ oxidation of phthalhydrazide 12 with lead tetraacetate[17] to the corresponding phthalazinedione 13 in the presence of excess cyclopentadiene, Scheme 5.
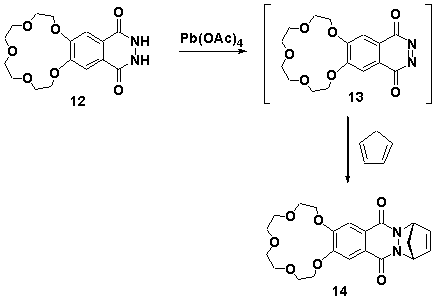
Scheme 5
A-BLOCK 14 was used to prepare the crown ether analogues of 5 and 7. Heating a mixture of 14 and cyclobutene epoxide 4 (sealed tube, dichloromethane, 140oC) yielded mono crown 15 in 43% yield, Scheme 6. The yield of this reaction was improved to 72% under photochemical conditions (acetone, 300nm, Rayonet reactor). Reaction of 14 with bis (cyclobutene epoxide) 6 under thermal conditions (sealed tube, dichloromethane, 140oC) was successful for the synthesis of bis (crown ether) 16 in 48% yield, Scheme 6. The spectral data for both 15 and 16 confirmed that similar stereoselective coupling had occurred.
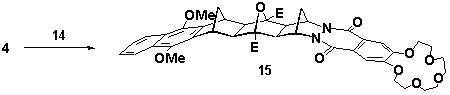
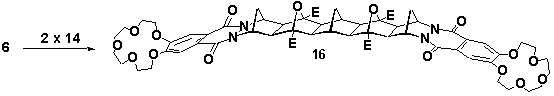
Scheme 6 E=CO2Me
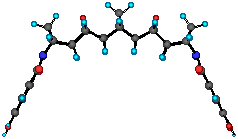
Figure 1 Geometrical Optimization of 16 at AM1 Level. The crown ether and ester moieties have been omitted.
Molecular modelling has been used to evaluate the shape of the bis (crown ether) 16 , Figure 1. The geometrical optimization of 16 was conducted at AM1 level with the crown ether and ester moieties removed to simplify the calculation. It is clear that the curvature of the [5]polynorbornane subunit significantly affects the geometrical interrelationship of the crown units. In this optimization the angles around the nitrogen atoms were constrained (average angle 120o) in accordance with known crystallographic data for similar systems.[16] This assumption finds experimental support from the adduct formed from the reaction of crown phthalazinedione 13 with anthracene 17 , Scheme 7.[18] Here the adduct 18 exhibits C2v symmetry in the NMR as required for a product with sp2 nitrogens.
Scheme 7
In conclusion, we have demonstrated that the cyclopentadiene adducts of phthalazinediones act as carriers (delivery agents) for crown ethers. The use of these A-BLOCKs under ACE coupling conditions allows entry to ribbon systems containing crown ethers rigidly linked to themselves or secondary effectors in geometrically precise structures.
4a,10a-Diaza -1,4-methano-1,4,4a,10a-tetrahydroanthrazine-9,10-dione 3.
Phthalhydrazide (1.62g, 10mmol) was suspended in a mixture of dichloromethane (50mL) and acetic acid (1.0mL) and cyclopentadiene (3.30g, 50mmol) added. Lead tetraacetate (4.43g, 10mmol) was added portionwise over 30 min with vigorous stirring and the mixture stirred for a further 30min before the resultant white precipitate was filtered and washed with dichloromethane (100mL). The combined filtrate and washings was evaporated and the residue recrystallized from methanol to yield A-BLOCK 3 (1.96g, 87%) as a white powder, m.pt. 246-8oC.
1H NMR (300MHz, CDCl3) d 2.08(1H, d, J=8.8Hz); 2.18(1H, dt, J=8.8, J=1.7Hz); 5.93-5.94(2H, m); 6.72-6.73(2H, m); 7.76-7.79(2H, m); 8.27-8.30(2H, m).
13C NMR (75MHz, CDCl3) d 49.38, 63.11, 127.09, 129.43, 132.91, 135.81, 155.19.
LRMS (CI) m/z (rel int) 226(M+, 59), 148(16), 130(17), 104(94), 76(69), 44(100).
HRMS (EI) C13H10N2O2 requires 226.0742, found 226.0741.
Synthesis of 5 via Thermal ACE
Cyclobutene epoxide 4 (15mg, 0.036mmol) and A-BLOCK 3 (8mg, 0.036mmol) in dichloromethane (0.5mL) was heated at 140oC for 4h in a teflon reaction vessel. Evaporation of the solvent and crystallization of the residue from methanol gave ACE adduct 5 (12mg, 55%) as an off-white powder, m.pt. 283-6oC.
1H NMR (300MHz, CDCl3) d 1.48(1H, d, J=9.7Hz); 1.90(1H, d, J=9.4Hz); 2.39(2H, s); 2.72(2H, d, J=9.4Hz); 2.78(2H, s); 3.00(1H, d, J=9.7Hz); 3.71(2H, s); 3.95(6H,s); 4.06(6H, s); 5.20(2H, s); 7.43-7.46(2H, m); 7.75-7.78(2H, m); 8.04-8.07(2H, m); 8.24-8.27(2H, m).
13C NMR (75MHz, CDCl3)d 41.91, 42.76, 52.91, 53.72, 54.99, 59.33, 61.30, 87.51, 122.16, 125.61, 127.33, 128.07, 129.59, 133.23, 133.59, 144.29, 153.69, 167.57
LRMS (ES) C36H32N2O9 requires 636; Found (M+1+) 637
Synthesis of 5 via Photochemical ACE Reaction
Cyclobutene epoxide 4 (13mg, 0.013mmol) and A-BLOCK 3 (7mg, 0.013mmol) were dissolved in acetone-d6 (1mL) in a quartz NMR tube, and irradiated at 300nm in the Rayonett apparatus. The reaction was monitored by 1H NMR and then stopped after complete disappearance of the starting compounds. The acetone was evaporated and the residue recrystallized from methanol to yield ACE adduct 5 (16mg, 80%) as an off-white powder, m.pt. 286-90oC.
The spectral data of the adduct from this reaction was identical with the product from the thermal ACE reaction.
Synthesis of 7 via Thermal ACE Reaction
Bis (cyclobutene epoxide) 6 (83mg, 0.20mmol) and A-BLOCK 3 (92mg, 0.41mmol) were dissolved dichloromethane (0.5mL) and heated at 140oC for 4h in a teflon reaction vessel. The crude product was passed through a short silica column and concentrated to give adduct 7 (110mg, 64%) as a grey powder, m.pt.>300oC.
1H NMR (300MHz, CDCl3) d 1.78(2H, d, J=10.8Hz); 1.92(2H, s); 1.95(4H,s); 2.17(2H,s); 2.72(4H,s); 2.87(2H, d, J=10.8Hz); 3.94(12H, s); 5.11(4H,s); 7.78(4H,m); 8.26(4H,m).
13C NMR (100MHz, CDCl3) d 33.97, 40.66, 52.87, 53.64, 54.53, 59.25, 75.99, 87.83, 127.40, 129.66, 133.27, 153.61, 167.29.
4,5-Bis(chloromethyl)benzo-15-crown-5 9
Hydrogen chloride gas was bubbled through a solution of formaldehyde (40%, 40mL) and hydrochloric acid (concentrated, 100mL) for 10min. To this mixture, a solution of benzo-15-crown-5 (4.288g, 16mmol) in hydrochloric acid (concentrated, 200mL) was slowly added over 2h, while hydrogen chloride gas was bubbled through the solution. The gas addition was continued for a further 15min. Then the solution was extracted with methylene chloride and the combined organic layer was dried over magnesium sulphate and the evaporation of the solvent afforded 9 (5.84g, quant.) as a white powder, m,pt. 134-5oC.
1H NMR (300MHz, CDCl3) d3.74(8H, m); 3.90(4H, m); 4.15(4H, m); 4.67(4H, s); 6.87(2H, s).
13C NMR (75MHz, CDCl3) d 43.29, 69.11, 69.38, 70.43, 71.03, 116.03, 129.09, 149.39.
LRMS (CI) m/z (rel int) 368(M+, 2x37Cl, 1), 366(6), 364(9), 329(2), 232(5), 197(100).
4,5-Bis(acetoxymethyl)benzo-15-crown-5 10
A mixture of 4,5-bis(chloromethyl)benzo-15-crown-5 9 (5.84g, 16.0mmol) and sodium acetate (2.67g, 32.5mmol) in glacial acetic acid (200mL) was refluxed for 20h. The acetic acid was removed in vacuo, water (100mL) added to the resultant solid and the aqueous suspension extracted with dichloromethane (3 x 100mL). The combined organic layers was washed with saturated sodium bicarbonate, water and dried (anhyd. sodium sulfate). Removal of the solvent in vacuo yielded 10 (6.10g, 91%) as a crude yellow solid. The product was used in the next reaction without further purification, m.pt. 73-4oC.
1H NMR (300MHz, CDCl3)d 2.07 (6H, s); 3.73 (8H, m); 3.89 (4H, m); 4.15 (4H, m); 5.11 (4H, s); 6.91 (2H, s).
13C NMR (75MHz, CDCl3) d 20.92, 63.52, 69.27, 69.51, 70.54, 71,11, 116.24, 127.81, 149.15, 170.63.
LRMS (CI) m/z (rel int) 412(M+, 4), 352(2), 310(4), 222(4), 178(37), 43(100)
4,5-(1,4,7,10,13-Pentaoxacyclopentadecano)phthalic anhydride 11
Crown ether 10 (5.0g, 12.1mmol) in sodium hydroxide (6M, 100mL) was refluxed for 2d. The reaction mixture was cooled to 80oC and a saturated solution of potassium permanganate (5.56g, 35.2mmol) in water added portionwise until the resultant green colour persisted for 5-10min. The reaction mixture was kept at 80oC for 1h, cooled and the precipitate removed via filtration (supercell). The resultant brown solution was acidified with concentrated hydrochloric acid to yield a yellow solution. The solvent was removed in vacuo to yield a light brown solid. The solid was taken up in acetic anhydride (100mL) and heated to 120oC for 6h. The solution was cooled and sodium chloride removed via filtration. The solvent was removed in vacuo to yield crude (29) (2.953g, 72%) as a dark brown solid. Compound 11 was used in the following reaction with further purification, m.pt. 154-6oC.
1H NMR (300MHz, CDCl3) d 3.77 (8H, m); 3.95 (4H, m); 4.25 (4H, m); 7.26 (2H).
LRMS (CI) m/z (rel int) 336(M+-2, 36), 207(32), 162(20), 113(48), 60(100).
6,7-(1,4,7,10-Pentaoxapentadecano)-2,3-dihydrophthalazine-1,4-dione 12
A solution of crown ether 11 (2.2g, 6.50mmol) and hydrazine monohydrate (0.38mL, 7.80mmol) in glacial acetic acid (100mL) was refluxed for 4h. The reaction mixture was cooled and solvent removed in vacuo to yield the crude product. Recrystallisation from methanol yielded 12 (1.298g, 57%) as a white solid, m.pt. 352oC.
1H NMR (300MHz, DMSO-D6) d 3.74 (8H, m); 3.92 (4H); 4.30 (4H, m); 7.50 (2H, s).
13C NMR (100MHz, DMSO-D6) d 68.33, 69.38, 70.51, 106.41, 121.83, 152.23, 165.39
LRMS (CI) m/z (rel int) 352(M+, 16), 221(18), 220(79), 162(37), 134(11), 113(12), 73(11), 44(100).
HRMS (EI) C16H20N2O7 requires 352.1270, found 352.1266.
4a,10a-Diaza-6,7(1',4',7',10',13'-pentaoxacyclopentadecano)-1,4,4a,10a-tetrahydroanthrazine-9,10-dione 14
6,7-(1,4,7,10,13-Pentaoxacyclopentadecano)-2,3-dihydrophtahlazine-1,4-dione 12 (352mg, 1mmol) and freshly distilled cyclopentadiene (330mg, 5mmol) were mixed in dichloromethane (20mL) and lead tetraacetate (443mg, 1mmol) added portionwise over 0.5h. The mixture was stirred for another 10min and then filtered through a short column of alumina. The solution was concentrated in vacuo and the excess cyclopentadiene removed under a stream of nitrogen. The residue was crystalized from methanol to yield 14 (332mg, 80%) as a white powder, m.pt. 264-8oC.
1H NMR (300MHz, CDCl3) d 2.05(1H, d, J=8.7Hz); 2.16(1H, d, J=8.7Hz); 3.76(8H,m); 3.93(4H,m); 4.26(4H,m); 5.91(2H,m); 6.71(2H,m); 7.26(2H,s)
13C NMR (75MHz, CDCl3) d 49.65, 63.30, 68.80, 68.99, 70.19, 71.24, 108.99, 123.91, 135.94, 153.23, 155.36
LRMS (CI) m/z (rel int) 416(M+, 91), 294(22), 206(24), 205(29), 163(21), 162(41), 134(69), 45(100)
Synthesis of Crown Ether 15 via Thermal ACE Reaction
4a,10a-Diaza-6,7(1',4',7',10',13'-pentaoxacyclopentadecano)-1,4,4a,10a-tetrahydroanthrazine-9,10-dione (20mg, 0.048mmol) and cyclobutene epoxide 4 (20mg, 0.48mmol) were dissolved in dichloromethane (1.00mL) and heated at 140oC for 4hrs in a teflon reactor. The solvent was removed under reduced pressure and the residue crystallized from methanol to yield 15 (17mg, 43%) as an off-white powder, m.pt. 280-82oC.
1H NMR (300MHz, CDCl3)d 1.37(1H, d, J=9.8Hz); 1.80(1H,d, J=9.8Hz); 2.32(2H,s); 2.63(1H, d, J=10.5Hz); 2.68(2H,s); 2.90(1H,d, J=10.5Hz); 3.62-3.69(8H,m); 3.84-3.90(4H,m); 3.87(6H,s); 3.98(6H,s); 4.15-4.20(4H,m); 5.10(2H,s); 7.36(m); 7.48(2H,s); 7.97(2H, m).
LRMS (ES) C44H46N2O14 requires 826; Found 825.
Synthesis of Crown Ether 15 via Photochemical ACE Reaction
4a,10a-Diaza-6,7(1',4',7',10',13'-pentaoxacyclopentadecano)-1,4,4a,10a-tetrahydroanthrazine-9,10-dione (20mg, 0.048mmol) and cyclobutene epoxide 4 (20mg, 0.48mmol) were dissolved in acetone-d6 (1.00mL) in a quartz NMR tube and irradiated at 300nm in the Rayonett apparatus. The reaction was monitored by 1H NMR until complete (6h). The solvent was removed under a stream of nitrogen and the residue was crystallized from methanol to yield 15 (32mg, 72%) as an off-white powder, m.pt. 280-84oC.
The spectral data of the adduct from this reaction was identical with the product from the thermal ACE reaction.
Synthesis of Bis (Crown Ether) (16 via Thermal ACE Reaction
4a,10a-Diaza-6,7(1',4',7',10',13'-pentaoxacyclopentadecano)-1,4,4a,10a-tetrahydroanthrazine-9,10-dione 14 (66mg, 0.158mmol) and bis (cyclobutene epoxide) 6 (33mg, 0.0793mmol) were dissolved dichloromethane (2mL) in a teflon reaction vessel and heated at 140oC for 2h. The solvent was removed under reduced pressure and the residue purified by column chromatography (10% methanol in ethyl acetate) to yield 16 (47mg, 48%) as a brown powder, m.pt >350oC.
1
H NMR (300MHz, CDCl3) d 1.77(2H, d, J=11.5Hz); 1.89(4H, s); 2.15(2H, s); 2.64(2H, s); 2.67(4H, s); 2.82(2H, d, J=11.5Hz); 3.69(16H, m); 3.89(8H, m); 3.91(12H, s); 4.21-4.25(8H, m); 5.05(4H, s); 7.52(4H, s).LRMS (ES) C61H68N4O24 requires 1240, found 1240.
Diels-Alder Reaction of Phthalazinedione 13 and Anthracene 17
6,7-(1,4,7,10,13-Pentaoxacyclopentadecano)-2,3-dihydrophtahlazine-1,4-dione 12 (80mg, 0.227mmol) and anthracene (41mg, 0.23mmol) were mixed in dichloromethane (10mL) containing two drops of glacial acetic acid and lead tetraacetate (100mg, 0.227mmol) added portionwise over 0.5h. The mixture was stirred at room temperature for 10min and then filtered through a short column of neutral alumina and concentrated in vacuo to yield 18 (69mg, 57%) as a white powder, m.pt. 246-50oC.
1H NMR (300MHz, CDCl3) d 3.67(8H,m); 3.84(4H,m); 4.16(4H,m); 7.19(4H, m); 7.30(2H, s); 7.48(4H, m); 7.52(2H,s).
13C NMR (75MHz, CDCl3) d 57.18, 68.72, 68.94, 70.15, 71.21, 109.03, 122.98, 124.13, 128.82, 134.07, 138.56, 153.29
LRMS (CI) m/z (rel int) 528(M+, 8), 294(8), 179(16), 178(100), 149(25), 85(16), 83(22).
AA thanks the University of Colombo for a leave of absence. Funding support from the Central Queensland University URG Scheme is gratefully acknowledged. We thank Dr W. S. Wilson for pioneering work with phthalazinediones.[19]
1. Pedersen, C. J. J. Am. Chem. Soc. 1967, 89, 7017; For a comprehensive review on this subject see also: Bradshaw, J. S.; Izatt, R. M.; Bordunov, A. V.; Zhu, C. Y.; Hathaway, J. K., Chapter 2: Crown ethers, Gokel, G. W., Vol. Ed.; in the series Comprehensive Supermolecular Chemistry; Lehn, J.-M.; Atwood, J. L.; Davies, J. E. D.; MacNicol, D. D.; Vogtle, F., Series Ed.; Pergammon: New York, 1996; Vol. 1, pp35-95.
2. Beer, P. D. J. Chem. Soc. Chem. Commun. 1986, 1678.
3. Beer, P. D.; Crane, C. G.; Drew, M. G. B. J. Chem. Soc. Dalton Trans. 1991, 12, 3235.
4. Voyer, N.; Deschenes, D.; Bernier, J.; Roby, J. J. Chem. Soc. Chem. Commun. 1992, 134.
5. Fages, F.; Desvergne, J.-P.; Bouas-Laurent, H.; Lehn, J.-M.; Barrens, Y.; Marsau, P.; Meyer, M.; Albrecht-Gary, A.-M. J. Org. Chem. 1994, 59, 5264.
6. Beer, P. D.; Stokes, S. E. Polyhedron 1995, 14, 2631.
7. Otsuki, J.; Russell, K. C.; Lehn, J.-M. Bull. Chem. Soc. Jpn 1997, 70, 671.
8. Thuery, P.; Nierlich, M.; Bryan, J. C.; Lamare, V.; Dozol, J.-F.; Asfari, Z.; Vicens, J. J. Chem. Soc. Dalton Trans. 1997, 22, 4191.
9. Warrener, R. N.; Wang, S.; Russell, R. A.; Gunter, M. J. Synlett, 1997, 47.
10. Redman, J. E.; Beer, P. D.; Dent, S. W.; Drew, M. G. B. Chem. Commun. 1998, 231.
11. Warrener, R. N. in "Advances in Strain in Organic Chemistry", Vol 6, Ed B. Halton, JAI Press Inc, pp 97-138; Fringuelli, F. and Taticchi, A. in "Dienes in the Diels-Alder Reaction", John Wiley and Sons, Inc, New York, 1990.
12. Boger, D. L.; Weinreb, S. N. in "Hetero Diels-Alder Methodology in Organic Synthesis", Academic Press, Inc, San Diego, 1987.
13. Warrener, R. N.; Schultz, A. C.; Butler, D. N.; Wang, S.; Mahadevan, I. B.; Russell, R. A.; Chem. Commun. 1997, 1023.
14. Warrener, R. N.; Margetic, D.; Russell, R. A. Synlett, 1998, in press.
15. 7-oxa and 7-isopropylidene norbornenes are rare exceptions and they give primarily endo,exo-stereoisomers together with the extended-frame products.
16. Agmon, I.; Kaftory, M.; Nelson, S. F.; Blackstock, S. C. J. Am. Chem. Soc., 1986, 108, 4477.
17. Clement, R. J. Org. Chem. 1960, 25, 1724.
18. The adduct of 3 with anthracene has been described. Clement, R. J. Org. Chem. 1962, 27, 1115.
19. Wilson, W. S. PhD Thesis, Australian National University, 1971.