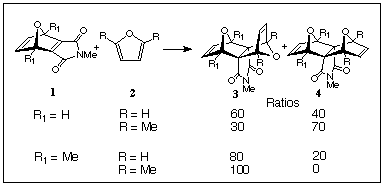
A continuing theme of our present research on the development of new molecular architectures, has been to explore cycloaddition stereoselectivies in the reactions of cyclic 1,3-dienes with norbornenes and related dienophiles since the control of product ring-junction stereochemistry is essential to the design of such architectures. In this regard, we have previously studied the reaction of furans 2 with 7-oxanorbornadienomaleimides (ONM) 1[1] where we have found that stereoselectivities of the adducts 3 and 4 could be influenced by introducing substituents at the 2,5-positions of the furans or at the bridgeheads of the 7-oxanorbornadienomaleimides (see product ratios - Scheme 1).
Further, the stereoselectivities of the examples studied were
predicted without exception using AM1 calculations to assess
the energies of the competing transition states leading to adduct
formation. We proposed that repulsive orbital-orbital interaction
between the oxygen bridge on the dienophile and the oxygen of
the furan was a significant factor in affecting the stereochemical
outcome. Similar orbital-orbital repulsions can also be invoked
to explain the loss of stereoselectivity of 7-oxabenzonorbornadiene
5 in its reaction with cyclobutene epoxide 7 (dipolar
ACE coupling [2]), in which adducts 9 (extended-frame product) + 10 (bent-frame product) were produced.
This result contrasts with the benzonorbornadiene 6 (X=CH2)
addition to the same epoxide dipole generator 7 where
only the extended-frame product 8 was observed.
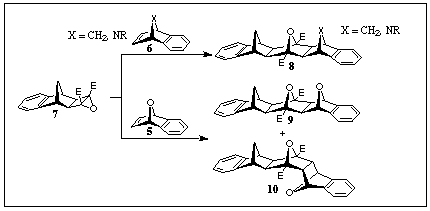
Again, 7-azabenzonorbornadienes 6 (X=NR) produced only
the extended-frame adduct 8 (X=NR) [3] with the dipole
from cyclobutene epoxide 7, thereby contrasting with their
oxygen counterparts 5 but behaving analogously with the
carbon-bridged benzonorbornadienes 6 (X=CH2).
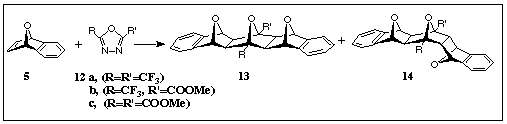
Similar patterns of non-selective additions involving oxygen-oxygen repulsions were observed in the products, 13 and 14, of the related dipolar couplings of a series of 2,5-bis-substituted oxadiazoles 12a,b,c to 7-oxabenzonorbornadiene 5 (Scheme 3) [4].
Since our recent focus in this area has been on 7-azanorbornyl
systems [3], accordingly, we initiated the present
experimental and theoretical study to investigate this topic with
regard to the stereoselectivity of the addition of N-substituted
pyrroles [5] and isoindoles to 7-oxanorbornadienomaleimides.
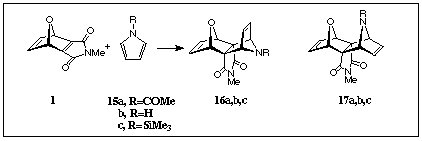
7-Oxanorbornadienomaleimide (ONM) 1 (R1=H) is
a very reactive, transient dienophile which is available from
the N-methyldibromomaleimide/furan Diels-Alder adduct [1] by treatment
with freshly made zinc/silver couple in tetrahydrofuran. When
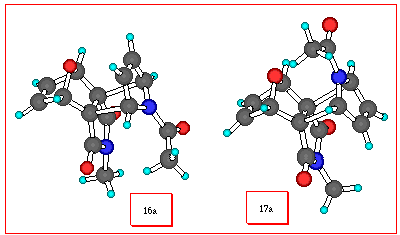
generated in the presence of N-substituted pyrrole 15a
in a refluxing THF solution, a single adduct 17a is formed
in modest yield, but with no evidence for the isomeric adduct
16a being formed. The proton chemical shift of the N-Me
group of the cyclic imide of the adduct (d
= 2.72 ppm) is considered diagnostic [1] for the structure given
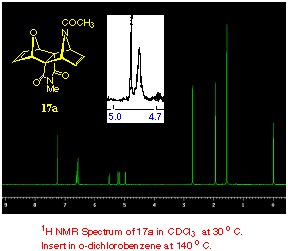
being shielded by the proximity of the newly formed p-bond (see Figure 1 below). The locked configuration of the N-acetyl group of 17a at room temperature, is clearly seen in the
proton NMR spectrum (see Experimental) where the asymmetric environment
of the four bridgehead protons (H1, H3, H6, H8) results in all of
them having well resolved individual resonances each integrating
for one proton. Upon heating to 140 oC (o-dichlorobenzene
solvent), these signals collapse to a pair of two proton signals
as expected (1H NMR of 17a). Such behaviour involving hindered rotation of amides is, of course, a well recognized phenomenon [6] involving the thermally sensitive restriction of rotation around the -CO-N- bond.
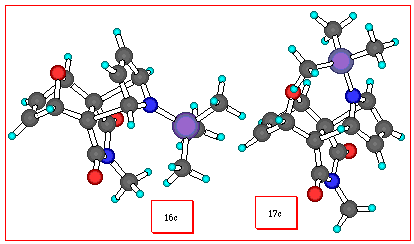
In the presence of free pyrrole 15b, no adduct was observed
when 1 (R1=H) was generated under the
same conditions as above. However, when N-trimethylsilylpyrrole
15c was used in a similar addition, the adduct 16c of
opposite stereochemistry to that formed from N-acetylpyrrole
was the only product observed, albeit again in moderate yield (Scheme 4) (1H NMR of 16b).
In this third case of this series, the actual product isolated was the desilylated material 16b arising from hydrolysis during the work-up of the adduct 16c. The stereochemical outcome of this experiment was founded on the chemical shift of the N-Me group of the cyclic imide at d 2.87 ppm. A comparison of the chemical shifts of the N-Me groups for these and similar related pairs of extended/bent-frame adduct isomers [7] is shown in Figure 1. In each of the four isomeric pairs, that isomer having the newly formed olefin proximate to the N-methyl group (ie the extended-frame isomer) consistantly shows a shielded proton chemical shift for its N-methyl resonance. The chemical shift differences Dd in ppm, for these pairs are also shown.
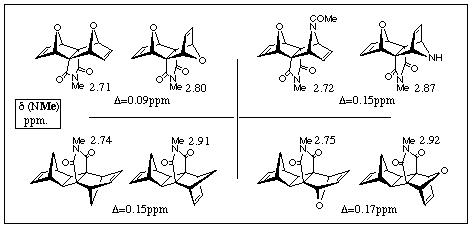
Preliminary studies by proton NMR spectroscopy of the related
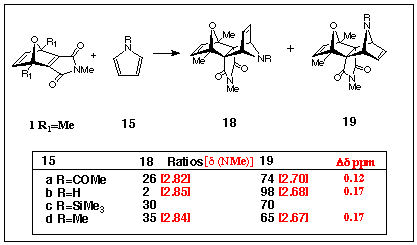
additions of the dienophile ONM1 (R1=Me)
with the four pyrroles 15a,b,c,d show that in all
cases, mixtures of [4+2] cycloadducts form (see Scheme
5). Thus the addition of ONM1 (R1=Me) with N-acetyl
pyrrole 15a, led to a mixture of the bent- and extended-frame
isomers 18a/19a in 26/74 ratio with the respective
chemical shifts of the imide N-methyl groups at d 2.82 and 2.70 ppm. Surprisingly (since no adduct was found for the 1 (R1=H) attempted cycloaddition), ONM
1 (R1=Me) did give an adduct with unsubstituted
pyrrole 15b and this was assigned the extended-frame structure
19b (d N-Me(imide) =
2.68ppm). A very small quantity of the bent-frame isomer 18b
was also detected. With trimethylsilylpyrrole 15c, a 30:70
mixture of adducts 18b and 19b was observed, the
TMS groups being once again displaced by hydrogen during the work-up
proceedure. The respective chemical shifts for the N-methyl groups
attached to the imide nitrogen atom of 18b and 19b
were d 2.85ppm and d2.68ppm. Finally, in the ONM1 (R1=Me) series, cycloaddition of N-methylpyrrole 15d again provided a mixture of adducts 18d/19d in 35:65 ratio with the imide N-methyl group chemical shifts at d 2.84 and 2.67 ppm respectively. These data are collected in Scheme 5 and although full experimental details are still being collected,
it is clear that the chemical shifts of the N-methyl groups of
the extended-frame isomers 19a-d again are all shielded when compared
with those of their respective bent-frame isomers 18a-d. It is also
important to note that the bridgehead methyl substituents in dienophile ONM
1 (R1=Me) dramatically alter the isomeric ratios
of the adducts. In all the pyrrole 15a-d additions, the
extended-frame isomers 19a-d predominated but (except for the parent
pyrrole15b), significant bent-frame components 18
were detected and this should be contrasted with the reversal
of stereochemistry observed in cycloadditions of the dienophile ONM
1(R1=H) noted above.
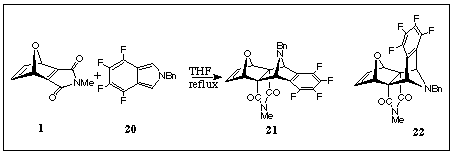
For an example of the corresponding cycloaddition of the dienophile ONM
1 (R=H) with an isoindole, the stable isoindole 20 [8] was chosen. Reaction between these addends in refluxing tetrahydrofuran produced the extended-frame adduct 21 (Scheme 6) in 21% yield with no evidence for the bent-frame system 22 being found in this case.
A complete application of high level ab initio methods is generally prohibitive for any but relatively small systems. However, semiempirical procedures applicable to the study of larger molecular systems have been very successfully applied to numerous chemical problems. We have shown previously that semiempirical calculations can be used to succesfully predict the stereochemical outcomes of cycloaddition reactions [1,9].
Although semiemprical methods generally overestimate and ab initio method underestimate activation barriers, relative activation energies are much less sensitive to the level of theory employed and generally provide reliable reactivity trends.
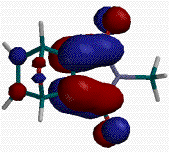
For the case at hand, cycloaddition site selectivity (ie addition to one or the other of the two nonequivalent double bonds in 1 (R1=H)) can be easily predicted by examination of frontier molecular orbitals of 1 (R1=H). The LUMO of 1 (R1=H) has much larger coefficients on the imide p system, with just a small contribution from the unsubstituted double bond (Figure 2).
Transition state calculations using the PM3 method [10] for the model reaction of the exo face of ONM 1 (R1=H) with pyrrole 15b, predict the favourable formation of exo,exo- product 17b. Adduct 17b is prefered by 3.4 kcal/mol over the bent-frame adduct 16b. Furthermore, the two possible adducts originating from the pyrrole attack onto the endo- side of the norbonadienomaleimide p bond have much larger energy barriers. These endo,endo- and endo,exo- products have respectively 8.3 and 6.2 kcal/mol larger activation energies than 17b, confirming the observed p facial selectivity.
For the reaction of the exo face of ONM 1 with N-acetylpyrrole 15a, the PM3 method predicts that the extended-frame adduct 17a is favoured over the bent-frame isomer 16a by 2.6 kcal/mol, in full accordance with the experimental observation. The activation energies were also estimated using ab initio 3-21G and 6-31G* basis sets by single point calculations using the PM3 transition state structures (see Figure 3). Although the activation energies are smaller, the relative energy differences are larger and follow the same trend, predicting isomer 17a to be preferred over 16a by 7.4 and 5.1 kcal/mol (3-21G//PM3 and 6-31G*//PM3 levels, respectively).
Our calculations further show that the substitution of pyrrole nitrogen with a trimethylsilyl group reverses the stereochemical outcome. For the reaction of the exo face of 1 with N-trimethylsilylpyrrole 15c, the PM3 method predicts that the bent-frame adduct 16c is favoured over the extended-frame isomer 17c (see Figure 4), as observed experimentally. The single point ab initio energy calculations also predict that the isomer 16c is preferred over 17c at both 3-21G//PM3 and 6-31G*//PM3 levels.
The calculations relating to the transition states for the additions
to the dienophile 1(R1=Me) are in progress.
We are grateful to the Australian Research Council (ARC) and the
Central Queensland University (URG)) for financial support of
this work. Dr. I.G.Pitt is also thanked for his early contributions
to the chemistry of dienophiles of type 1.
(1a,2b,3a,6a,7b,8a)N-Methyl-11-Acetyl-14-methyl-12-aza-11-oxatetracyclo-[6.2.1.13,6.02,7]dodeca-4,9-diene-2,7-dicarboximide
(17a).
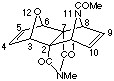
Freshly made Zn/Ag couple (500 mg) was added to a solution of 2,6-dibromo-4-methyl-4-aza-10-oxa-tricyclo(5.2.1.02,6)dec-8-ene-3,5-dione
[1] (337mg, 1.0 mmol) and acetylpyrrole (110 mg, 1.0 mmol)
in dry THF (4 ml) and the mixture was heated to refux for 1 h.
The solvent was evaporated and the residue chromatographed
(silica gel, ether/petrol ether (40-60 oC) 1:1) to give
the crude product 17a which was recrystallized from EtOAc
(42 mg, 21%), mp 209-210 oC.
1H NMR (30 oC. CDCl3) d 1.95
(3H, s), 2.72 (3H, s), 4.98 (1H, s), 5.16 (1H, s), 5.23 (1H,
s), 5.51 (1H, s). 6.56-6.63 (4H, m). 13C NMR
(CDCl3) d 21.14,
24.54, 59.22, 62.70, 68.51, 69.04, 80.97, 81.29, 138.83, 138.96,
139.44, 139.84, 165.38, 173.96, 174.14. LRMS m/z 286 (M+,
1.5%), 218 (35.8), 109 (12.2), 67 (100), 43 (73.5); HRMS m/z 286.0953
(C15H14N2O4 = 286.0953).
(1a,2a,3b,6b,7a,8a)N-Methyl-11-Acetyl-11-aza-12-oxatetracyclo-[6.2.1.13,6.02,7]dodeca-4,9-diene-2,7-dicarboximide
(16b).
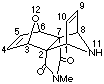
Freshly made Zn/Ag couple (250 mg) was added to a solution of 2,6-dibromo-4-methyl-4-aza-10-oxa-tricyclo(5.2.1.02,6)dec-8-ene-3,5-dione[1](10mg, 0.30mmol) and N -trimethylsilylpyrrole (200 mg, 1.4 mmol) in dry THF (4 ml). The mixture was refluxed for 2 h, cooled, treated with water (30 ml) and then extracted
with dichloromethane (2 x 20 ml ).The extract was dried (Na2SO4), evaporated, and the residue chromatographed (silica gel, Et2O) to give the adduct which was recrystallized from EtOAc to afford pure 16b (8 mg, 10%), mp 136-138 oC.
1H NMR NMR(CDCl3)
d 2.87 (3H, s, NMe), 4.75 (2H, t, J=0.7Hz,
H1,8), 5.15 (2H, t, J=0.9 Hz, H3,6), 6.52 (2H, t, J=0.9 Hz, H4,5),
6.55 (2H, t, J=0.7 Hz, H9.10). 13C NMR (CDCl3)
d 25.62 (NMe), 69.87 (C2,7), 78.10
(C1,8), 82.15 (C3,6), 135.83 (C4,5), 139.05 (C9,10), 176.87 (carbonyl).
LRMS m/z 244 (M+, 0.5%), 216 (30.2), 188 (23.6), 177 (6.2), 176
(0.6), 68 (100).
(1a,2b,3a,6a,7b,8a)N-Methyl-15-Benzyl-10,11,12,13-tetrafluoro-15-aza-16-oxapentacyclo-[6.6.1.13,6.02,7.09,14]hexadeca-4,9,11,13-tetraene-2,7-dicarboximide
(19).
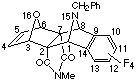
Freshly made Zn/Ag couple was added to a solution of 2,6-dibromo-4-methy-4-aza-10-oxa-tricyclo(5.2.1.02,6)dec-8-ene-3,5-dione
1 (50mg, 0.15 mmol) and 9-benzyl-5,6,7,8-tetrafluoroisoindole
18 (31 mg, 0.15 mmol) in dry THF (2 ml). This mixture was
heated to refux for 1 h and the solvent was then evaporated. The
residue was subjected to chromatography (silica gel, Et2O/PE
1:1) and to provide the crude product 19 which was recrystallized
from EtOAc (12 mg, 21%), mp 208-209 oC. 1H
NMR (CDCl3) d 2.39
(3H,s), 3.40 (2H, s), 4.93 (2H, s), 5.32 (2H, s), 6.57 (2H, s),
7.26-7.32 (5H, m). 13C NMR (CDCl3)
d 24.96, 52.96, 67.47, 68.40, 82.11,
126.35, 128.33128.89, 129.43, 138.06. 139.43, 146.10, 174.29.
LRMS m/z 456 (M+, 0.8%), 388 (24.6), 279 (15.7), 177 (2.5), 91
(100), 68 (16.0). HRMS m/z 456.1099 (C24H16F4N2O3 =456.1097).
† Visiting Research Fellow, Centre for Molecular Architecture, CQU, 1996,1998; Department of Chemistry,University of Leicester, LE1 7RH, UK.
1. R.N.Warrener, G.M.Elsey, L.Maksimovic, M.R.Johnston, C.H.L.Kennard; Tetrahedron Lett. 1995, 36, 7753-7756.
2. R.N.Warrener, A.C.Schultz, D.N.Butler, S.Wang, I.B.Mahadevan, R.A.Russell; Chem. Commun., 1997, 1023-1024.
3. D.N.Butler, J.R.Malpass, D.Margetic, R.A.Russell, R.N.Warrener; Synlett. 1998, in press.
4. V.G.Seitz, H.Wassmuth; Chemiker Zeitung, 1998, 118, 80; R.N.Warrener, D. Margetic, unpublished results.
5. Z.Chen, M.L.Trudell; Chem. Rev. 1996, 96, 1179-1193.
6. M.G.B.Drew, A.V.George, N.S.Isaacs; J. Chem. Soc. Perkin Trans. 1, 1985, 1277-1284.
7. R.N.Warrener,I.G.Pitt, D.N.Butler; unpublished results.
8. G.M.Priestley, R.N.Warrener; Tetrahedron Lett., 1972, 4295-4298.
9. for example see; R. N. Warrener, R. A. Russell, D. Margetic, Synlett, 1997, 38-40; R. N. Warrener, D. Margetic, E. R.Tiekink, R. A. Russell, Synlett, 1997, 196-198; R. N. Warrener,G. M. Elsey, M. A. Houghton, Tetrahedron Lett. 1995, 36, 1417-1418.
10. J. J. P. Stewart J. Computat. Chem. 1989, 10, 209 - 220 and J. J. P. Stewart J. Computat. Chem. 1989, 10, 221 - 264.
{kind=link}
{kind=link}