The following text format files can be read in directly with MacMolPlt via the Open and Append menus. MacMolPlt will automatically determine the type of file by looking first for a file extension. If it finds .PDB or .XYZ then it treats the files as Protein Data Bank and XMol XYZ files. If these extenstions are not found then it searchs the file for certain phrases such as "GAMESS version" for log files, "IRC information packet" for IRC files and "NATOMS=" on the first line of .mol files.
MacMolPlt is capable of exporting to several file formats. Most export formats are accessed through the Export Menu item. This menu command will bring up a file dialog allowing you to name the target file and choose the export format.
GAMESS log file means any type of GAMESS log file. MacMolPlt reads geometry points, energies, frequencies (if any), orbitals, and other related information. MacMolPlt can only read IRC points out of log files created with the April 20, 1995 version and later (previous versions did not write out a full set of coordinates). You may append log files similarly to IRC files (see IRC notes below). Beginning with version 3.0 MacMolPlt reads in the basis set (meaning coefficients and exponents) and molecular orbital vectors (including localized orbitals if present). This has the effect of increasing the file size when the log file is saved. Each frame is searched for orbitals, but GAMESS does not normally punch orbitals to the log file for points other that the first and last point of a file. If you wish to view orbitals for other points you can either Import the $Vec group from the .Dat file or set the NPRT=1 option in either the $STATPT gourp or the $IRC group to force GAMESS to punch orbitals for every point. This will not permit computing a total electron density for MCSCF wavefunctions since GAMESS does not print the MCSCF natural orbitals and occupation numbers except for the first and last points. Note: MacMolPlt looks for certain phrases in the file to locate each piece of information, thus I do not advise editing log files as you might prevent MacMolPlt from correctly reading the file.
GAMESS log files containing effective fragment coordinates are also now supported. For log files with frequency information and fragments you must have a GAMESS version from September 1997 or later which prints frequency information for fragments just like any other atom (instead of translations and torques). Note: the January 6, 1998 version of GAMESS changed the way optimizations print out coordinates. Thus you will need MacMolPlt 4.6 or later to read the new style of output.
GAMESS IRC (usually with a .irc extension) are punched out by GAMESS for any IRC or DRC type run. These files contain a minimum amount of information about each geometry point and are thus the most compact way to store the IRC or DRC information. MacMolPlt can read in all types of IRC files and keeps the coordinates, energy, IRC distance or DRC time. You may append further geometry points from other files. This feature is designed to make it easy to piece together multiple pieces of an IRC or DRC. Geometry points will be ordered according to the IRC distance or DRC time regardless of their position in the files. Any duplicate points (duplicate means equal IRC distance or DRC time) will be skipped. On IRC's you may wish to change the sign of the IRC points on one side of the Transition state. To make flipping the sign easier there is now an option to automatically flip the sign when a file is opened. To use this option simply check the box in the open file dialog. If you wish to piece together multiple IRC's from multiple transition states you will need to offset the second and subsequent transition states from the first (defined as point 0). To offset the points simply type in the offset distance in the edit field of the Open dialog. To calculate the offset distance add the maximum IRC distance for the forward IRC from the first TS to the maximum IRC distance for the IRC in the reverse direction from the second TS. These features should make it easy to create a seamless IRC from one minimum to another.
MacMolPlt reads ASCII data files in a method similar to, but not the same as the free format input method used in MOLPLT. Free format input allows you to enter data without regard to what column it is in or what case it is. A blank space, a tab, or an equals sign can be used to separate data fields. An example of a keyword input item is DELTA=0.05, which is two data items, the first (DELTA) being the keyword telling what the data is, the second (0.05) being the actual value. Because you can give keywords in any order you like, "DELTA=0.05 NOPLOTS" and "NOPLOTS DELTA=0.05" are the same. In other circumstances, you will be expected to input a set of data items without a keyword. You must give these data items in the order expected by the program. If a read site expects an integer and you give a floating point number, it will be truncated. If the read site expects a floating point number, you can give it in several forms: 10.0, +10.0, 10, and 1.0E1 are all floating point tens. If you give less data than expected, an error will occur (in other words, trailing zeros in "3 0 0 0"will not be filled in if you just type "3").
Part 1: NATOMS=<natoms> [NBONDS=0] [BNDLENGTH=2.0] [MODE=0] [NKINDS=<nkinds>]
Part 2: <title>
Part 3:
Part 4: <atom> <x> <y> <z> [<dx> <dy> <dz>]
Part 5: BONDATOMS <iatom> <jatom>
Part 6:
A sample .mol file is:
Example input
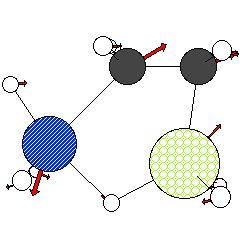
NATOMS= 14 NKINDS= 4 NBONDS= 14 MODE=-1 TiH2ethSiH4-Transition State #1 freq=-122.8 C 1 .30 TI 8 .51 H 5 .23 SI 6 .48 C 1.337665 1.279412 0.001392 0.22066 0.06268 0.00967 C -0.124205 1.362164 -0.001731 0.28546 0.13557 -0.00664 Ti 0.934914 -0.714544 -0.000306 0.07972 0.07543 -0.00194 Si -1.848838 -0.163114 0.000363 -0.07748 -0.19922 0.00070 H 1.846560 1.585021 -0.899870 0.29077 -0.06884 0.00590 H 1.842378 1.584093 0.905346 0.26746 -0.08094 0.03085 H -0.548065 1.798772 -0.891198 0.19759 -0.00337 -0.02735 H -0.551623 1.800156 0.885339 0.17598 -0.00445 0.0043 H -2.323342 -0.813825 -1.242937 0.33602 -0.17918 0.08437 H -2.598203 1.124432 -0.004376 0.31162 0.01070 -0.02841 H -2.314463 -0.802227 1.252755 -0.25060 -0.09815 -0.00814 H -0.634063 -1.467347 -0.000927 -0.14727 0.12827 -0.02397 H 1.658771 -1.326577 -1.446235 -0.28643 -0.05401 -0.05408 H 1.656909 -1.328383 1.445790 -0.30601 -0.06866 0.05343 BONDATOMS 2 1 3 1 3 12 4 9 5 1 6 1 > 7 2 8 2 13 3 4 10 4 11 3 14 4 12 > 2 4
This might result in the following image (depending on color preferences and atom sizes):
MacMolPlt now has a very simple minded ability to read in PDB files. What this means is that MacMolPlt will read in most (all?) PDB files and will read in all (including duplicates and HETATM's) atoms as generic atoms (ie. C, N, O instead of Ca, Cb, etc...). No connection or other special information will be read, just the basic atom types and positions. If you don't want to see duplicate atoms or HETATM's then you will have delete them by hand before reading the file...
Once you have read in molecule with a basis set (usually from a GAMESS log file) you may use the Import menu item to read $VEC groups from the associated .Dat file. MacMolPlt can not determine which $VEC group goes with which frame or even if the $VEC group goes with the current molecule. Thus when importing a $VEC group you will be presented with a list of the $VEC groups found in the file and asked to choose the one to import to the current frame. Note that the file does not have to be a .dat file punched by GAMESS, it can be any text file containing a complete $VEC group.
There are a couple of ways to create GAMESS input format files. The first, but limited method, is to use the Export menu item and choose the GAMESS $DATA group format. This will punch the coordinates for the current (or all) frame out to a text file using the GAMESS COORD=CART format. This can be handy if you want to run single points on a series of geometries from an IRC or DRC.
The second method is to use the MacMolPlt input builder. The input builder allows you to choose many GAMESS options and then, by clicking the Write File button, write a GAMESS input file out to disk. The input builder is far from complete, but does allow you to create simple GAMESS input files quite easily. One nice feature is that if you define a set of internal coordinates in the Coordinates window and set the number of Z-matrix variables correctly MacMolPlt will automatically punch out a $ZMAT group.
MacMolPlt can not currently read in GAMESS input files.
One of the features of QuickDraw 3D is a cross platform 3D metafile format for storing all QuickDraw 3D object data. If QuickDraw 3D is active for a file you may choose the 3MDF format in the export dialog. This will create a text 3DMF file which can be read by any program which supports the 3DMF format (for example SimpleText). You may also place these files on the WWW in combination with a Netscape plugin such as wulrplug. See the QuickDraw 3D information for more information.
If you have the QuickTime library installed and have either normal modes or multiple frames available then you will have the option of exporting to a QuickTime Movie. The resulting movie will be stored at the size of target window and will be both temporaly and spatially compressed as well as flattened. Thus the file will be fairly small and can be put on WWW sites without modification. At this point you can not change the compression quality, which is set to the "normal" values. Frame animations have the additional option of including an energy plot appended to the right side of the molecule animation.
If you have QuickTime installed you can export any image into a JPEG image file. QuickTime is required because I use the compressor built into QuickTime to create the JPEG. The image is based on the target window size at 72 dpi, thus it is best suited to creating still images for WWW pages. The compression settings are not user modifiable and are set to some reasonable "normal" quality values.
To aid users in copying QuickDraw 3D images into other applications you can export the target window into a hi-res (360 dpi) PICT file. Then you can import the PICT into other apps such as ChemDraw or WordPerfect. Note: you can copy PICTs out of a target window directly using the copy command. This works fine for 2D images as they copy out as vector graphics at 300 dpi, but due to the size of QuickDraw 3D bitmap images you only get 72 dpi images which do not look very good when printed. Note: Creating the large PICT may require a lot of memory depending on the size of the window.
MacMolPlt binary files contain the internal data format of MacMolPlt. Saving to a binary file preserves all data and all preferences. These files are designed to reduce disk space requirements for storing large amounts of information and reduce the time required to open files. MacMolPlt will read binary files written by any previous version of MacMolPlt, however you should not expect older versions of MacMolPlt to read files written by newer versions.
MacMolPlt can read and write (via the export menu) XMol style XYZ files. The XYZ format is a very simple format for storing molecular structures and can have multiple frames. The basic format is:
1st line MUST contain the number of atoms
1 line label
One line for each atom given in the format:
H 1.0 1.0 1.0
If you want multiple frames simple concatinate the data from individual frame together with no intervening blank lines or other data. The format also allows for display of one normal mode. To do so simple append the offset vector to the end of each atom line. An example file with two frames and an optional normal mode is:
3 Frame 1 CH2 C 0.0000 0.0000 -0.0899 H -0.8929 0.0000 0.5353 H 0.8929 0.0000 0.5353 3 Frame 2 H2O O 0.0000 0.0000 0.0372 0.0000 0.0000 0.2644 H 0.0000 -0.7581 -0.5986 0.0000 0.4331 -0.5267 H 0.0000 0.7581 -0.5986 0.0000 -0.4331 -0.5267
MacMolPlt implements a simple format for importing and exporting 2D and 3D surface data. To export a surface first create a surface and make sure that the surface is updated so that the grid data is available for export. Then choose the Export menu command and the Surface export format. MacMolPlt will then export the surface grid data to the file you specify (this is useful to check the input format as well). To import a 2D or 3D surface create a new General 2D or 3D surface. From the surface dialog you can then choose the file containing the grid data. The format for the 2D and 3D files are similar but there are a couple of differences. Floating point numbers may be given in a variety of formats and the grid data may contain any number of values per line.
For 2D planes the format is:
1Line Label
30 # grid points
-4.165882 -4.306789 0.839681 //Origin of the 2D grid
0.297019 0.000000 0.000000 //X inc vector
0.000000 0.297019 0.000000 //Y inc vector
-0.000018 -0.000021... (# of grid points squared grid values)
For 3D surfaces:
1 Line label
35 12 41 //nx ny nz
-4.687880 -1.587537 -5.059790 //Origin of the 3D grid
0.280144 0.288642 0.275207 //x increment, y inc, z inc/
grid(x(y(z)))
0.000000 0.000000... (nx*ny*nz grid values)