You will be using the Silicon Graphics workstations in the Perkin laboratory
and you will need your valid e-mail user identification and password. When you initiate MacroModel from the
"Applications menu", you are prompted by a request for the type of device you are using. Type 2. This is
followed by a number of "windows" which will appear in the screen. The
largest is the molecule drawing area, and to the right of it are the main
command windows. In addition, a number of menus "pop-up" in response to certain
commands.
The mouse buttons invoke different actions on these various windows.
- The left mouse button is used to "focus" a window, ie establish communication
between it and the keyboard from which file names etc can be typed, and to
select individual menu items.
- The middle button is used to rotate the molecule in the drawing window. The
sense of rotation is selected from the main button windows by clicking on ROT X
(or Y or Z). By default, both ROT X and ROT Y are selected, and moving the
mouse whilst holding down the middle button will rotate the molecule about
these two axes. To reset the default, "double click" on ROT X.
- The right hand button is used to bring help. For example, clicking with this
button on a menu item opens up the Help window, with suitable comments. If more
text than fits the size of this window is present, use the "scroll" bars and
the left hand button to view the rest of the text;
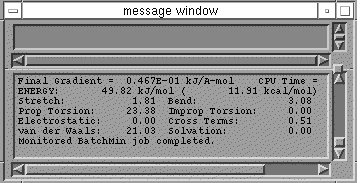
One window always present is the message window. Here are reported numerical
results of various calculations and other information of relevance.
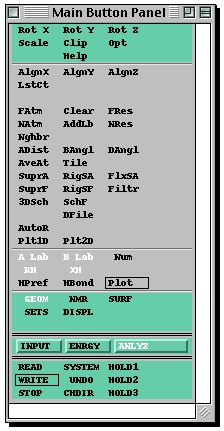
The most important menus are the three shown below. You switch from one to the
other by clicking with the left button on INPUT, ENRGY, or ANLYZ, and you will
need to use them in that order. You will only need to use a few of the menu
items, but feel free to experiment with the others!


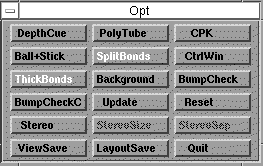
An options menu is invoked by clicking on OPT in any of the three main menus.
You can switch ie CPK models on at any time, but be aware it slows things down
considerably;
- To start a carbon chain, click DRAW in INPUT and click on the molecule window
with the left mouse button to start a chain. You can rotate the structure
whilst building by pressing the middle mouse button and "dragging" the mouse
around. In this way, you can impart a realistic 3D component to the molecule
you are building. Pressing the right mouse button enables you to "translate"
the molecule without rotating it.
- There are some organic "templates" defined on the INPUT menu. If these are
more appropriate, select one of these instead of DRAW and position it in the
middle of the screen. Other templates can be introduced by selecting PEPTID,
NUCLEI or CARBO from the menu.
- To continue building up a chain, move the cursor and press the left button
again. A line will appear on the screen, representing a C-C bond. If you have a
template to which you want to attach a chain, select DRAW, and position the
cursor at the atom you want the chain added to. This may create a pentavalent
carbon, and you will have to use H DEL (see below) to delete eg a
hydrogen and H ADD to add them. If you click once on these two menu items, the
action performed will be on a single atom only, but if you double-click, a new
menu pops up with several options;

Quite a number of menu items are of this kind.
- Continue building up the chain. If you want to close to a cyclic compound,
position the cursor accurately on the first atom. If you want a double bond,
back-track to the previously positioned atom in the chain.
- One frequently needs to break the chain, ie to lift the imaginary pen
you are drawing with off the paper, such as when returning to a previously
defined single bond to turn it into a double or triple bond. To do this, simply
select DRAW again. To enter carbon side-chains, first select DRAW, then point
to the carbon carrying the side chain and then start building up a new chain.
After this chain is complete, the "pen" must be lifted off the paper as
described above. Complete all the basic skeleton of the molecule in this
manner, representing all heteroatoms by carbon at this stage.
- To substitute any carbon atom by a heteroatom, left click on the heteroatom
in the INPUT menu, then point to the carbon to be substituted. If you want to
insert say four oxygen atoms, point to O on the menu, then to each of the four
carbon atoms in turn.
- To delete a specified atom, click on DELET once, then point to the atom to be
deleted, or to the middle of a bond if you want to change a double into a
single bond. Double click to get options;
To remove a single bond, you have to first turn it into a "zero order" bond
(used for weak complexes, transition states etc) and then delete the zero order
bond.
- If the structure you are drawing is too big for the screen, it can be
rescaled by selecting the SCALE option. Clicking SCALE twice rescales to
exactly the size of the screen. Alternatively, if you want to magnify part of
structure, select CLIP and point the cross-hair at the bottom left and top
right of the region of the molecule to be magnified.
- Carbon chains so drawn have no hydrogens, whereas templates do. Very probably
at this stage, the carbon valencies are not correct, most missing hydrogen and
the templates may have too many. To ensure correct valency, double click on H
DEL and then on H ADD to ensure the correct valencies. If these are wrong, the
program will not minimise the structure correctly.
- When the basic elements of the structure are complete, select the ENRGY menu.
Several default options are selected automatically, including the MM2 force
field. Click on another force field if you want tho change this option. Select
START to commence geometry optimisation. You will be asked to enter a COM file
name. If you are performing a calculation for the first time, select New. If
you have such a file from a previous run, select Old. In the former selection,
you will have to type the name of a file. Notice that the keyboard is only
active for this purpose if the cursor is positioned over the rectangular box in
which the file name is entered.

From now on, information about the state of the optimisation appears in the
message window. If that window is obscured (e.g. by the Options window) you may
have to move it by pressing the left mouse button on the top bar of the window
and "dragging" the menu to a new location..The geometry on the screen will be
updated as the optimisation proceeds, giving some idea of how the shape of the
molecule is changing. If your 3D sketching has not given the program a good
enough indication of the three-dimensional structure of the molecule, the
optimisation may not converge to a sensible answer. Alternatively, your
starting point may converge to the wrong conformation (such as boat rather than
chair in a cyclohexane ring).You have to select the INPUT option to make
appropriate alterations, and minimise again. Several cycles of this may be
needed before a satisfactory result is obtained. You should particularly check
that you have the correct stereochemistry. For example, if you think you are
working with a cis-alkene, make sure the program has not rearranged it to a
trans-alkene when you were not looking! There are also some 'local minima' in
the MM2 or 3 potential energy surface which have very high energies. If an
energy makes no 'chemical sense', you should look very carefully at the
geometry.
- When optimisation is complete, you are now ready to calculate parameters such
as bond lengths, angles etc. Select ANALYZ and use ADIST, BANGL or DANGL to
obtain bond lengths, angles or torsion angles, or NMR to obtain NMR coupling
constants. Record all the energies in your laboratory notebook, since there
is no easy way of printing the values.
- Save your work using WRITE. Place the mouse cursor in the first
rectangular entry window (just below the Write caption) so that a
type cursor appears and type a name in from the keyboard. If the mouse cursor
is not positioned there, you will not be able to enter a name. Then click on
Write. If you wish to display these structures in other programs, you
might also want to write out a PDB (Brookhaven format) file by selecting
the button for this option. this will allow you to read such files using other
programs such as CAChe or MacMolPlt or Chem3D.
- If you are calculating a series of structures, it is easier to use the
previous structure as a template for the next, rather than starting from
scratch. However, there are some problems in doing this that you might
encounter. If you add a double bond to a structure with hydrogens in place, it
is possible to arrive at a pentavalent carbon atom. Make sure that you delete
any hydrogens associated with a double bond, and than add them all again with
the ADD H option. If in doubt, you can delete ALL hydrogens by selecting H DEL
and add using ADD H. If for whatever reason, you suspect an energy or a
structure you might have obtained might be too high, then redefine the molecule
and re-optimise. The "variation principle" applies, ie the lowest energy is the
most correct. It is also possible to get variation in final energies of +/- 5
kcal if the molecule has several possible conformations, since the minimisation
does not always find the lowest energy form. MacroModel in fact has some very
sophisticated options (Multi-conformer searching, Monte-Carlo minimisation,
Molecular dynamics) to overcome these problems, but these are beyond the scope
of the present technique.
- To obtain plots of your final optimised structures, select
the PDB option in the WRITE menu. The file will be written to what corresponds
to your network drive (Windows or Macintosh). It can be opened on these systems
using a variety of programs, including RasMac, WebLabViewer, MacMolPlt (Mac only),
JMol, MacSpartan (Mac only) etc. In the file navigation dialogs, you should select
this network drive first before attempting to open the file. Most of these
programs allow you to save an appropriate image of the molecule for inclusion in
your report. You can also try selecting the molecule, copying and seeing
if it pastes into Word or other word processor.
Back to Experiment
Macromodel Home page