Orbital Diagrams for Simple Molecules
Instructions for Viewing
Ensure your browser has a 3D model plugin installed; Quick3D or 3DMFPlugin (both browser plugins), 3DMF
Optimizer (a Macintosh application), Geo3D (for Mac) or 3DMF Viewer for Windows. Windows users must also
install the
QuickDraw3D libraries from Apple. To view a
model, click on the picture of any model. A separate window
will open containing the model.
Localised Orbitals in Methane and Cyclopropane
Note how the localised orbital in methane lies along the axis of the C-H bond, whereas
the localised orbital in cyclopropane does not lie along the axis of the C-C bond, but
is bent outside it (the banana shape, although in fact its more triangular than banana
shape).
The Bonding and Anti-bonding p
Molecular Orbitals in Ethene and Cyclopropane
Ethen and cyclopropane are classified as alkene and alkane respectively, but the latter
is often regarded as having properties associated with the former. These orbitals show why.
| Ethene: Bonding |
Ethene:Anti-bonding |
Cyclopropane: bonding |
Cyclopropane: Anti-bonding |
 |
 |
 |
 |
The Anti-bonding X-Y Molecular Orbital (LUMO) along the
series XH3-Y
Nucleophilic attack on haloalkanes is often presented in terms
of the highest occupied molecular orbital (HOMO) of the
nucleophile interacting with the lowest unoccupied molecular
orbital (LUMO) of the electrophile. This interaction is
favoured both by good overlap between these two orbitals and by
minimising the energy difference between them. To see how this works in
practice, the table below shows the quantitative form and
energies of the LUMO orbitals of the electrophile.
Various points are noteworthy. Thus although the LUMO of
fluoromethane has the appropriate shape for an SN2
displacement by a nucleophile, its energy is very high and
hence it does not interact at all favourably with the nucleophilic HOMO
(F is rarely displaced in SN2
reactions). In contrast, the LUMO of iodomethane has an
appropriate energy, but its shape has a higher density at the
iodine end than the carbon end. Thus e.g. I- can attack at the
iodine end under suitable circumstances. Finally, we note that
whilst substitution at carbon electrophiles always goes with
inversion, Silicon can substitute with retention of
configuration, as the shape and energy of its LUMO in SiH3Fimplies.
| Y |
X=Carbon |
X=Silicon |
X=Germanium |
X=Tin |
X=Lead |
| Y=Fluorine |
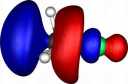 |
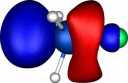 |
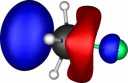 |
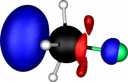 |
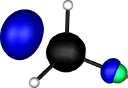 |
| Y=Chlorine |
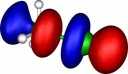 |
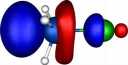 |
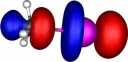
| Y=Bromine |
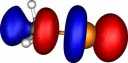 |
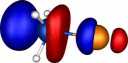 |
| Y=Iodine |
 |
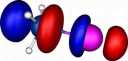 |
| Y=Astatine |
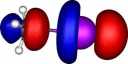 |
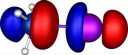 |
How were these orbitals produced?
Molecular Orbitals (MOs) are solutions of the Schroedinger
equation for a molecule. Each MO has an associated eigenvalue
(a discrete energy level) for an electron in a molecule, and is
described by a set of coefficients (the wavefunction) expressed
as a linear combination of the atomic orbital components of
each atom in the molecule. The Hartree-Fock method is
approximate way of solving the Schroedinger equation, a method
implemented in a Quantum computational program called GAMESS.
One particular representation of the atomic orbitals for each
molecule is selected (called the basis set, in our case the
3-21G* basis) and the Hartree-Fock equations solved. An eigenvalue solution
of these equations yields
molecule orbitals (also often called canonical or delocalised orbitals), which are rendered visible by calculating
the value of the wavefunction at any point in space and then
contouring these values at one particular level (in our case
0.06 Atomic units) to create an "iso-surface" using a program
called MacMolPlt. The result is written out into a 3D computer
graphics file in 3DMF format, and displayed in a browser using
a 3DMF plug-in. It is also possible to transform molecular canonical orbitals to
a more localised form (an Edmiston-Ruedenberg transform) which are the ones shown
above for methane and cyclopropane.

