The π-resonance in amides famously helped Pauling to his proposal of a helical structure for proteins. Here I explore some geometric properties of amides related to the C-N bond and the torsions about it.
Archive for the ‘crystal_structure_mining’ Category
π-Resonance in amides: a crystallographic reality check.
Saturday, September 5th, 2015Mesomeric resonance in substituted benzenes: a crystallographic reality check.
Wednesday, August 26th, 2015Previously, I showed how conjugation in dienes and diaryls can be visualised by inspecting bond lengths as a function of torsions. Here is another illustration, this time of the mesomeric resonance on a benzene ring induced by an electron donating substituent (an amino group) or an electron withdrawing substituent (cyano).
A visualisation of the effects of conjugation; dienes and biaryls.
Tuesday, August 25th, 2015Here is another exploration of simple chemical concepts using crystal structures. Consider a simple diene: how does the central C-C bond length respond to the torsion angle between the two C=C bonds?
Intermolecular atom-atom bonds in crystals? The O…O case.
Saturday, July 25th, 2015I recently followed this bloggers trail; link1 → link2 to arrive at this delightful short commentary on atom-atom bonds in crystals[1] by Jack Dunitz. Here he discusses that age-old question (to chemists), what is a bond? Even almost 100 years after Gilbert Lewis’ famous analysis,[2] we continue to ponder this question. Indeed, quite a debate on this topic broke out in a recent post here. My eye was caught by one example in Jack's article: "The close stacking of planar anions, as occurs in salts of croconic acid …far from producing a lowering of the crystal energy, this stacking interaction in itself leads to an increase by several thousand kJ mol−1 arising from Coulombic repulsion between the doubly negatively charged anions" I thought I might explore this point a bit further in this post.
References
- J.D. Dunitz, "Intermolecular atom–atom bonds in crystals?", IUCrJ, vol. 2, pp. 157-158, 2015. https://doi.org/10.1107/s2052252515002006
- G.N. Lewis, "THE ATOM AND THE MOLECULE.", Journal of the American Chemical Society, vol. 38, pp. 762-785, 1916. https://doi.org/10.1021/ja02261a002
The Bürgi–Dunitz angle revisited: a mystery?
Tuesday, May 12th, 2015The Bürgi–Dunitz angle is one of those memes that most students of organic chemistry remember. It hypothesizes the geometry of attack of a nucleophile on a trigonal unsaturated (sp2) carbon in a molecule such as ketone, aldehyde, ester, and amide carbonyl. Its value obviously depends on the exact system, but is generally taken to be in the range 105-107°. A very good test of this approach is to search the crystal structure database (this was how it was originally established[1]).
References
- H. B:urgi, J. Dunitz, J. Lehn, and G. Wipff, "Stereochemistry of reaction paths at carbonyl centres", Tetrahedron, vol. 30, pp. 1563-1572, 1974. https://doi.org/10.1016/s0040-4020(01)90678-7
A new way of exploring the directing influence of (electron donating) substituents on benzene.
Friday, April 17th, 2015The knowledge that substituents on a benzene ring direct an electrophile engaged in a ring substitution reaction according to whether they withdraw or donate electrons is very old.[1] Introductory organic chemistry tells us that electron donating substituents promote the ortho and para positions over the meta. Here I try to recover some of this information by searching crystal structures.
References
- H.E. Armstrong, "XXVIII.—An explanation of the laws which govern substitution in the case of benzenoid compounds", J. Chem. Soc., Trans., vol. 51, pp. 258-268, 1887. https://doi.org/10.1039/ct8875100258
Halogen bonds 4: The strongest (?) halogen bond.
Sunday, December 7th, 2014
Continuing my hunt, here is a candidate for a strong(est?) halogen bond, this time between Se and I.[1]. 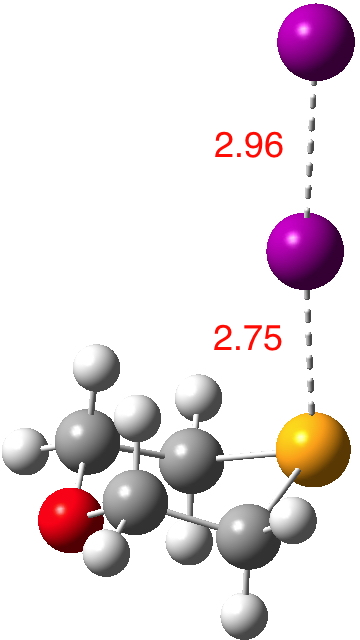 The features of interest include:
The features of interest include:
References
- H. Maddox, and J.D. McCullough, "The Crystal and Molecular Structure of the Iodine Complex of 1-Oxa-4-selenacyclohexane, C<sub>4</sub>H<sub>8</sub>OSe.I<sub>2</sub>", Inorganic Chemistry, vol. 5, pp. 522-526, 1966. https://doi.org/10.1021/ic50038a006
Halogen bonds: Part 1.
Saturday, November 29th, 2014Halogen bonds are less familiar cousins to hydrogen bonds. They are defined as non-covalent interactions (NCI) between a halogen atom (X, acting as a Lewis acid, in accepting electrons) and a Lewis base D donating electrons; D….X-A vs D…H-A. They are superficially surprising, since both D and X look like electron rich species. In fact the electron distribution around X-X (A=X) is highly anisotropic, with the electron rich distribution (the "donor") being in a torus encircling the bond, and an electron deficient region (the "acceptor") lying along the axis of the bond.
(more…)
Tags:crystal structure search, D. Note, frequent commentator, Paul Schleyer
Posted in crystal_structure_mining, Interesting chemistry, reaction mechanism | No Comments »