HCN is a weak acid (pKa +9.2, weaker than e.g. HF), although it does have an isomer, isocyanic acid or HNC (pka < +9.2 ?) which is simultaneously stronger and less stable. I conclude my halide acid series by investigating how many water molecules (in gas phase clusters) are required for ionisation of this “pseudo-halogen” acid.
How many water molecules does it take to ionise HI?
February 28th, 2015Why is this post orphaned from the previous? In order to have the opportunity of noting that treating iodine computationally can be a little different from the procedures used for F, Cl and Br.
How many water molecules does it take to ionise HF and HBr?
February 27th, 2015No doubt answers to the question posed in the previous post are already being obtained by experiment. Just in case that does not emerge in the next day or so, I offer a prediction here.
How many water molecules does it take to ionise HCl?
February 14th, 2015According to Guggemos, Slavicek and Kresin, about 5-6![1]. This is one of those simple ideas, which is probably quite tough to do experimentally. It involved blasting water vapour through a pinhole, adding HCl and measuring the dipole-moment induced deflection by an electric field. They found “evidence for a noticeable rise in the dipole moment occurring at n≈5–6“.
References
- N. Guggemos, P. Slavíček, and V.V. Kresin, "Electric Dipole Moments of Nanosolvated Acid Molecules in Water Clusters", Physical Review Letters, vol. 114, 2015. https://doi.org/10.1103/physrevlett.114.043401
How-open-is-it?
February 12th, 2015The title of this post refers to the site http://howopenisit.org/ which is in effect a license scraper for journal articles. In the past 2-3 years in the UK, we have been able to make use of grants to our university to pay publishers to convert our publications into Open Access (also called GOLD). I thought I might check out a few of my recent publications to see what http://howopenisit.org/ makes of them.
Chiroptical spectroscopy of the natural product Steganone.
February 10th, 2015Steganone is an unusual natural product, known for about 40 years now. The assignment of its absolute configurations makes for an interesting, on occasion rather confusing, and perhaps not entirely atypical story. I will start with the modern accepted stereochemical structure of this molecule, which comes in the form of two separately isolable atropisomers.

The first reported synthesis of this system in 1977 was racemic, and no stereochemistry is shown in the article (structure 2).[1] Three years later an “Asymmetric total synthesis of (-)steganone and revision of its absolute configuration” shows how the then accepted configuration (structure 1 in this article) needs to be revised to the enantiomer shown as structure 12 in the article[2] and matching the above representation. The system has continued to attract interest ever since[3],[4],[5],[6], not least because of the presence of axial chirality in the form of atropisomerism. Thus early on it was shown that the alternative atropisomer, the (aS,R,R) configuration initially emerges out of several syntheses, and has to be converted to the (aR,R,R) configuration by heating[3]. One could easily be fooled by such isomerism!
References
- D. Becker, L.R. Hughes, and R.A. Raphael, "Total synthesis of the antileukaemic lignan (±)-steganacin", J. Chem. Soc., Perkin Trans. 1, pp. 1674-1681, 1977. https://doi.org/10.1039/p19770001674
- J. Robin, O. Gringore, and E. Brown, "Asymmetric total synthesis of the antileukaemic lignan precursor (-)steganone and revision of its absolute configuration", Tetrahedron Letters, vol. 21, pp. 2709-2712, 1980. https://doi.org/10.1016/s0040-4039(00)78586-8
- E.R. Larson, and R.A. Raphael, "Synthesis of (–)-steganone", J. Chem. Soc., Perkin Trans. 1, pp. 521-525, 1982. https://doi.org/10.1039/p19820000521
- A. Bradley, W.B. Motherwell, and F. Ujjainwalla, "A concise approach towards the synthesis of steganone analogues", Chemical Communications, pp. 917-918, 1999. https://doi.org/10.1039/a900743a
- M. Uemura, A. Daimon, and Y. Hayashi, "An asymmetric synthesis of an axially chiral biaryl via an (arene)chromium complex: formal synthesis of (–)-steganone", J. Chem. Soc., Chem. Commun., vol. 0, pp. 1943-1944, 1995. https://doi.org/10.1039/c39950001943
- B. Yalcouye, S. Choppin, A. Panossian, F.R. Leroux, and F. Colobert, "A Concise Atroposelective Formal Synthesis of (–)‐Steganone", European Journal of Organic Chemistry, vol. 2014, pp. 6285-6294, 2014. https://doi.org/10.1002/ejoc.201402761
Mechanism of the solvatochromic reaction of a spiropyran.
February 4th, 2015The journal of chemical education has many little gems providing inspiration for laboratory experiments. Jonathan Piard reports one based on the reaction below[1]; here I investigate the mechanism of this transformation.
References
- J. Piard, "Influence of the Solvent on the Thermal Back Reaction of One Spiropyran", Journal of Chemical Education, vol. 91, pp. 2105-2111, 2014. https://doi.org/10.1021/ed4005003
Fine-tuning a (hydrogen) bond into symmetry.
January 23rd, 2015Sometimes you come across a bond in chemistry that just shouts at you. This happened to me in 1989[1] with the molecule shown below. Here is its story and, 26 years later, how I responded.
References
- P. Camilleri, C.A. Marby, B. Odell, H.S. Rzepa, R.N. Sheppard, J.J.P. Stewart, and D.J. Williams, "X-Ray crystallographic and NMR evidence for a uniquely strong OH ? N hydrogen bond in the solid state and solution", Journal of the Chemical Society, Chemical Communications, pp. 1722, 1989. https://doi.org/10.1039/c39890001722
A convincing example of the need for data repositories. FAIR Data.
January 15th, 2015Derek Lowe in his In the Pipeline blog is famed for spotting unusual claims in the literature and subjecting them to analysis. This one is entitled Odd Structures, Subjected to Powerful Computations. He looks at this image below, and finds the structures represented there might be a mistake, based on his considerable experience of these kinds of molecules. I expect he had a gut feeling within seconds of seeing the diagram.
The demographics of a blog readership – updated
January 8th, 2015About two years ago, I posted on the distribution of readership of this blog. The passage of time has increased this from 144 to 176 countries. There are apparently between 189-196 such, so not quite yet complete coverage! 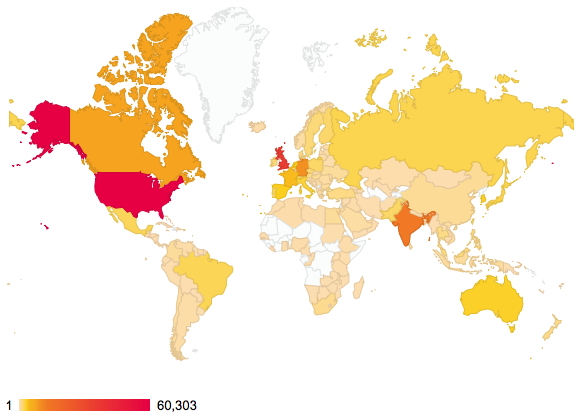
Of course, it is the nature of the beast that whilst we can track countries, very little else is known about such readerships. Is the readership young or old, student or professor, chemist or not (although I fancy the latter is less likely). Another way of keeping tabs on some of the activity are aggregators such as Chemical Blogspace, which has been rather quiet recently. Perhaps we have become too obsessed by metrics, and with the Internet-of-things apparently the “next-big-thing”, the metrics are only likely to increase. This will only encourage “game playing“, and I urge you to see a prime example of this in the UK REF (research excellence framework), the measure which attempts to rank UK universities in terms of their “excellence”.