December 29th, 2023
I will approach this example of a molecule-of-the-year candidate – in fact the eventual winner in the reader poll – from the point of view of data. Its a metallocene arranged in the form of a ring comprising 18 sub-units.[1] Big enough to deserve a 3D model rather than the static images you almost invariably get in journals (and C&EN). So how does one go to the journal and acquire the coordinates for such a model?
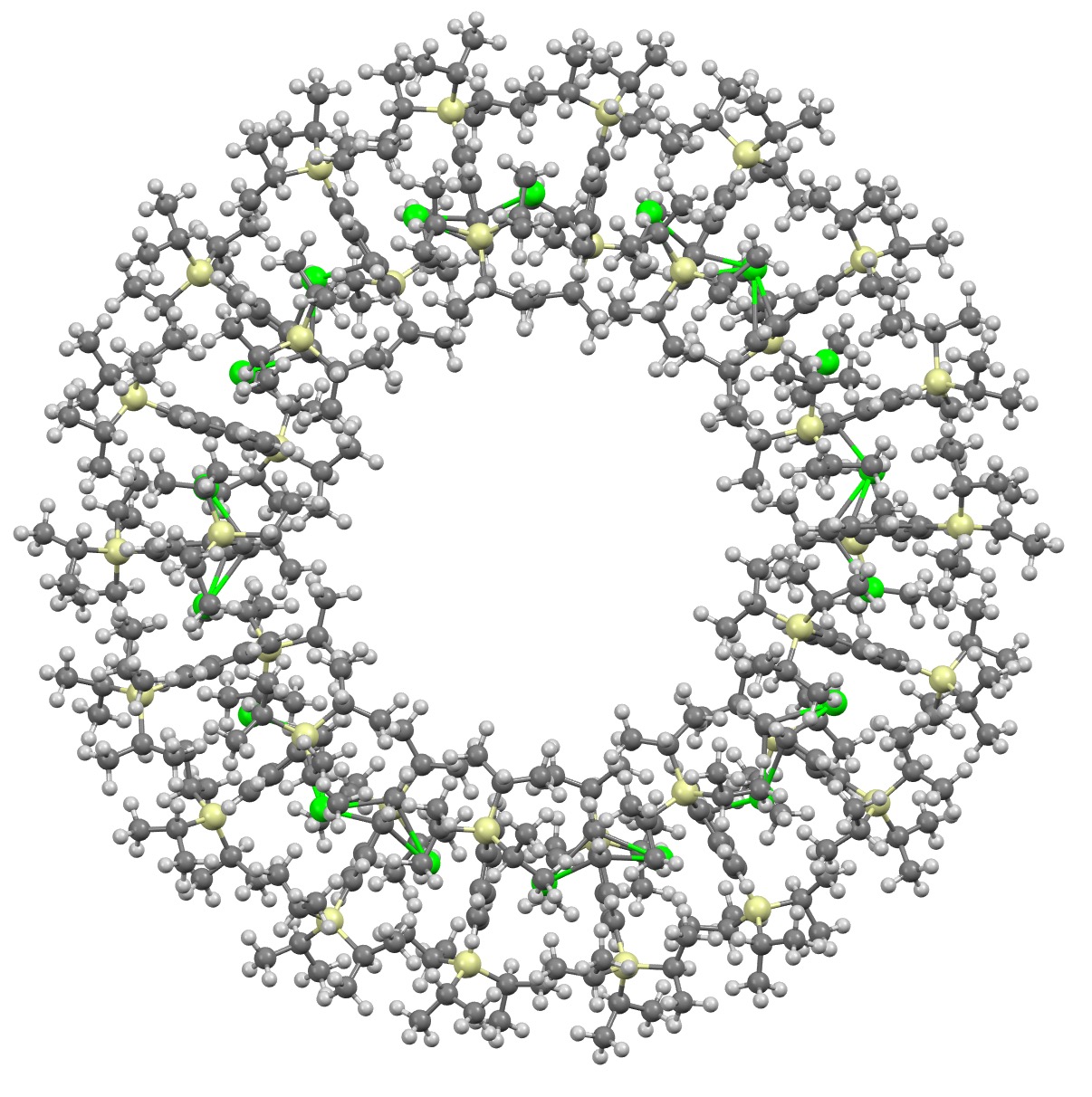
Read the rest of this entry »
References
- L. Münzfeld, S. Gillhuber, A. Hauser, S. Lebedkin, P. Hädinger, N.D. Knöfel, C. Zovko, M.T. Gamer, F. Weigend, M.M. Kappes, and P.W. Roesky, "Synthesis and properties of cyclic sandwich compounds", Nature, vol. 620, pp. 92-96, 2023. https://doi.org/10.1038/s41586-023-06192-4
Posted in Uncategorised | 3 Comments »
December 28th, 2023
Posted in Uncategorised | No Comments »
December 10th, 2023
Around 1996, journals started publishing what became known as “ESI” or electronic supporting information, alongside the articles themselves, as a mechanism for exposing the data associated with the research being reported and exploiting some of the new opportunities offered by the World Wide Web. From the outset, such ESI was expressed as a paginated Acrobat file, with the Web being merely a convenient document delivery mechanism. Such ESI would eventually reach more than 1000 such pages in length in some chemistry articles. The richer opportunities of Web interactivity were far less exploited. I have written about various aspects of this throughout this blog[1],[2],[3], together with one early compendium of our own data examples.[4] Here I update that compendium starting from 2005 to the current 2023 and add further information, being the current state of curation of some of these early examples. Curation became necessary because many of the earlier examples were no longer functional due to changes in the way journals expose these data objects or indeed changes at the data repository end of things over this 18 year period.
Read the rest of this entry »
References
- H. Rzepa, "Four stages in the evolution of interactive ESI as part of articles in chemistry journals.", 2022. https://doi.org/10.59350/qypm4-qfv97
- H. Rzepa, "Web page decay and Journals: How an interactive "ESI" from 2006 was rescued.", 2022. https://doi.org/10.59350/cqesx-a0e83
- H. Rzepa, "Curating a nine year old journal FAIR data table.", 2017. https://doi.org/10.59350/z9g5j-r2p69
- H. Rzepa, "(Hyper)activating the chemistry journal.", 2009. https://doi.org/10.59350/wczky-8sf79
Posted in Uncategorised | No Comments »
December 7th, 2023
In 2023, we very much take for granted that everyone and pretty much everything is online. But it was not always so and when I came across an old plan indicating how the chemistry department at Imperial College was connected in 1989, I was struck by how much has happened in the 34 years since. Nowadays all the infrastructures needed are effectively “built in” to the building when it is constructed and few are even aware of them. But in 1989 that was not at all true.
Read the rest of this entry »
Posted in Uncategorised | No Comments »
November 11th, 2023
I am a member of the Royal Society of Chemistry’s Historical group. Amongst other activities, it publishes two editions of a newsletter each year for its members. A new theme was recently launched asking for contributions on the topic of “two influential books” and shortly to appear in the winter 2023 edition will be the following recollections by myself (reprinted here with permission).
Read the rest of this entry »
Posted in Uncategorised | No Comments »
October 12th, 2023
In an earlier post on this topic,[1]‡ I described how the curly-arrows describing the mechanism of a nucleophilic addition at a carbonyl group choreograph in two distinct ways, as seen in red or blue below. The arrows in red can be described as firstly addition to the carbonyl group to form either a transient intermediate (a two-step process) or instead a formal transition state state as a concerted single-step mechanism. The blue arrows do the reverse; firstly elimination and then followed by addition. I will use the shorthand AE for the first type and EA for the second type. Here I explore some more nucleophiles to see which of these two mechanisms they follow. Data for these results can be found at 10.14469/hpc/13171
 N- carbon ylid: This is a very facile (low-barrier) reaction with a C-O bond length response that initially increases steeply, followed by a more modest decline and hence corresponds to an AE mechanism.
N- carbon ylid: This is a very facile (low-barrier) reaction with a C-O bond length response that initially increases steeply, followed by a more modest decline and hence corresponds to an AE mechanism.
Read the rest of this entry »
References
- H. Rzepa, "The "double-headed" curly arrow as used in mechanistic representations.", 2023. https://doi.org/10.59350/f00wf-5tq46
Tags: Interesting chemistry
Posted in Uncategorised | No Comments »
September 12th, 2023
Some 13 years ago, I speculated about the longevity of the type of science communication then (and still now) represented by Blogs. I noted one new project called ArchivePress that was looking into providing solutions equivalent to what scientific journals have done for some 350 years of science communication. The link to ArchivePress no longer works, but details of the project can still be found here. Since then the technology and infrastructure has moved on, with a new backbone provided by the use of persistent identifiers (PIDs) in the form of DOIs. The PID ecosystem is now extensive and so a revival of the concept has recently been launched called The Rogue Scholar. Here I take a look at some of is features and illustrate these with application to this blog.
Read the rest of this entry »
Posted in Uncategorised | 2 Comments »
August 29th, 2023
The schematic representation of a chemical reaction mechanism is often drawn using a palette of arrows connecting or annotating the various molecular structures involved. These can be selected from a chemical arrows palette, taken for this purpose from the commonly used structure drawing program Chemdraw. Explanations of how to apply the individual arrows are not always easy to find however! Circled in red are the ones to be discussed here, although most carry fascinating and often subtle meanings!‡

Read the rest of this entry »
Tags: Curly arrows
Posted in Curly arrows, reaction mechanism | No Comments »
August 25th, 2023
The Swern oxidation[1] is a class of “activated” dimethyl sulfoxide (DMSO) reaction in which the active species is a chlorodimethylsulfonium chloride salt. The mechanism of this transformation as shown in e.g. Wikipedia is illustrated below.‡ However, an interesting and important aspect of chemistry is not apparent in this schematic mechanism and to rectify this, a full computed mechanism is laid out below, for which the FAIR data has a DOI: 10.14469/hpc/13151
 Read the rest of this entry »
Read the rest of this entry »
References
- K. Omura, and D. Swern, "Oxidation of alcohols by “activated” dimethyl sulfoxide. a preparative, steric and mechanistic study", Tetrahedron, vol. 34, pp. 1651-1660, 1978. https://doi.org/10.1016/0040-4020(78)80197-5
Posted in Curly arrows, reaction mechanism | 3 Comments »