Mercury (IV) tetrafluoride attracted much interest when it was reported in 2007[1] as the first instance of the metal being induced to act as a proper transition element (utilising d-electrons for bonding) rather than a post-transition main group metal (utilising just s-electrons) for which the HgF2 dihalide would be more normal (“Is mercury now a transition element?”[2]). Perhaps this is the modern equivalent of transmutation! Well, now we have new speculation about how to induce the same sort of behaviour for caesium; might it form CsF3 (at high pressures) rather than the CsF we would be more familiar with.[3] Here I report some further calculations inspired by this report.
The argument goes something like this. Xenon difluoride (XeF2) is a well-known stable compound of xenon. Caesium comes immediately after xenon in the periodic table (electron shell properties [Xe].6s1) and so Cs+ would be iso-electronic with Xe. If the latter can form a stable difluoride (and higher), why not Cs? A neutral compound following this line of argument would therefore be CsF3.
So here comes a calculation. I used a large basis set (Def2-QZVPPD basis for Cs), with a pseudopotential describing 46 core electrons (including the two d-shells) and a further 8 in the 5s/p shell to make up the [Xe] core + 1 extra in the 6s shell) using the ωB97XD functional to obtain the geometry, shown below.[4]
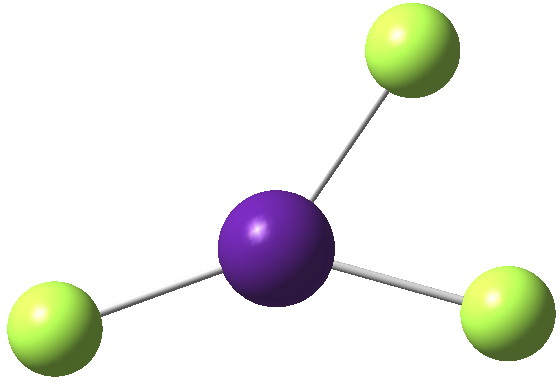
Click for 3D and normal modes
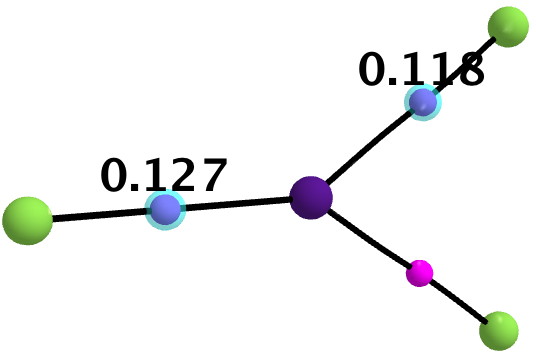
All 3N-6 normal vibrational modes are real, which indicates it is a proper minimum in the potential energy surface. A QTAIM analysis shows that ρ(r) at the bond critical points is pretty respectable for bonds (below). The QTAIM-derived average number of electrons on Cs is 7.4, which gives a charge of 1.6 on the Cs , thus involving the 5p shell as well as the 6s.
The most obvious question is what is the free energy change when the species dissociates to CsF and F2? This is exo-energic by 25.1 kcal/mol[5], not as large as you might have thought! Well, what about the barrier to such dissociation? The transition state for this process is shown below, delightfully asymmetric![6] and this gives a free energy barrier of 33.9 kcal/mol (the IRC smoothly defines this reaction[7],[8]).
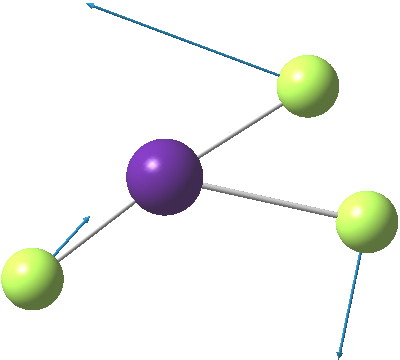
Click for animation
We may conclude from this brief foray that in CsF3 caesium would indeed deserve to be called a p-block element, although this is not quite as good as it sounds. Take a look for example at the highest occupied molecular orbital. It certainly involves the p-block, but this orbital is in fact anti-bonding, and the Cs-F bonds derive their strength from the lower occupied orbitals.
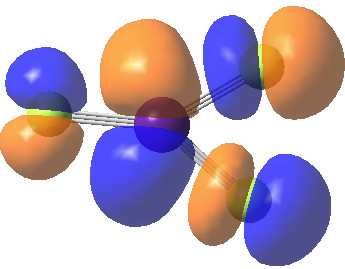
HOMO. Click for 3D
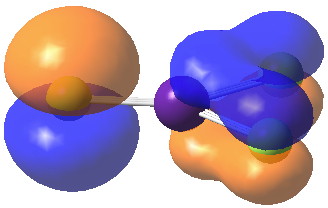
HOMO-6. Click for 3D
And the final take-home message. The report of this molecule[3] suggests it could be stable under high pressure. Here, the free energy barrier to dissociation is calculated to be indeed high, which implies that if made it could be kinetically quite stable even under normal pressures (in an inert matrix where it would be prevented from reacting with itself).
POSTSCRIPT: The LUMO below is also antibonding, but is mostly 6s on Cs. If one force-populates this by a double excitation from the HOMO, the resulting state is higher in energy (the wavefunction is stable to both singlet and triplet excitations). Which shows that Cs utilises (antibonding) 5p rather than (antibonding) 6s in this species.
Author
References
- X. Wang, L. Andrews, S. Riedel, and M. Kaupp, "Mercury Is a Transition Metal: The First Experimental Evidence for HgF<sub>4</sub>", Angewandte Chemie International Edition, vol. 46, pp. 8371-8375, 2007. https://doi.org/10.1002/anie.200703710
- M. Miao, "Caesium in high oxidation states and as a p-block element", Nature Chemistry, vol. 5, pp. 846-852, 2013. https://doi.org/10.1038/nchem.1754
- H.S. Rzepa, "Gaussian Job Archive for CsF3", 2013. https://doi.org/10.6084/m9.figshare.861029
- H.S. Rzepa, "Gaussian Job Archive for CsF3", 2013. https://doi.org/10.6084/m9.figshare.861030
- H.S. Rzepa, "Gaussian Job Archive for CsF3", 2013. https://doi.org/10.6084/m9.figshare.861038
- H.S. Rzepa, "Gaussian Job Archive for CsF3", 2013. https://doi.org/10.6084/m9.figshare.861047
Tags: animation, energy, free energy barrier, free energy change, metal, pence, post-transition main group metal, potential energy surface
What about Cs[F3] rather than Cs[F]3? Could fluorine form a trihalide anion instead? Could difluorine complexes be viable?
Good questions both. But the real issue is whether the dissociation barrier is going to be high or not.
Oh certainly it’s all about the dissociation barrier, but the question then becomes to what. Cs[F3] or a difluorine complex of CsF would seem decent alternatives to CsF + F2.
Disclaimer: I’m a first year student in the Netherlands and I have not received any formal training in computational chemistry.
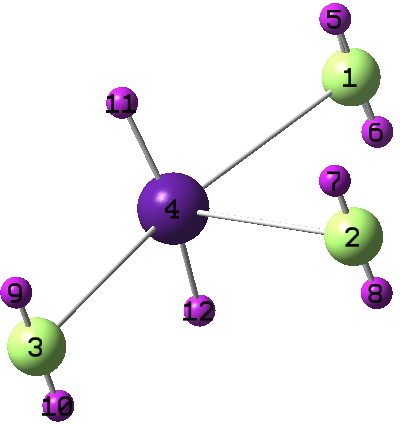
Here is the computed structure of the di-cation of CsF3 (computed since the HOMO shown above was anti-bonding). Indeed the bond orders of the di-cation are greater, as is ρ(r). It has the more familiar T-shape associated with eg ClF3.
This is a very interesting structure. Some remarks:
1) It seems to be anti-VSEPR since there are no visible lone pairs to explain the T-shaped structure. Then, is this really the global local minimum? Is there any other local minimum?
2) The amounts of electron density at the BCPs are larger than that of the neutral CsF3 ~0.12. What is mechanism for density “migration” to the BCPs in this dication? Seemingly the HOMO elimination somehow elevates the covalent nature of bonding. Are QTAIM localization (LI) and delocalization (DI) indexes available for these two species? I guess the DI must increase in the dication whereas the “percent” LI must diminishes. Are these really the case?
3) Seemingly, the dication is isoelectronic with IF3. Then, what is the structure of IF3? The iodine has two lone pairs…
4) Is is possible to add another fluorine atom to the dication without disturbing significantly its electronic structure? What about a dianion that makes the whole complex neutral?
5) Are there (3, -3) critical points in the core part of Laplacian of electron density of Cesium in dication? I guess there must be some to act like lone pairs. This has been observed some time ago by Ron Gillespie and Richard Bader for transition metal complexes and I remember a review paper by Robinson and Gillespie published in Angew. Chem. Int. Ed. on these exotic features of the Laplacian of electron density that act as “non-visible” lone pairs. However, I am not aware of such observations on Alkaline metals. Is this the first case?
1. I showed QTAIM. This does not routinely reveal lone pairs. A technique such as ELF is needed to reveal those, and indeed interesting they often are!
2. The electron density is higher because the bonds are much shorter.
3. IF3 (or ClF3) are very similar. The link above to ClF3 does show an ELF analysis of the lone pairs, and interesting they indeed were!
4. The dianion is in fact the original neutral CsF3 (ie CsF3(2+) + 2e = CsF3). The geometry changes as a result (the T-shaped dication becomes a transition state for pseudorotation of the neutral species). Yes, CsF5 and perhaps even CsF7 are both interesting species.
5. Yes, you can extract the lone pairs by various means; I use ELF to do so. The Laplacian is also interesting (I have discussed the Laplacian in various other posts here). A [3,-3] QTAIM point is the equivalent of a disynaptic basin, but you need monosynaptic basins for lone pairs.
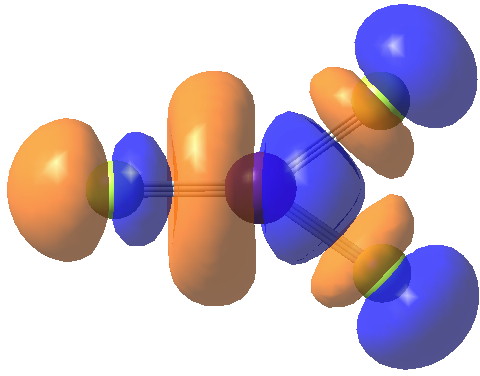
The ELF for CsF3 (2+) is shown below
As you can see, there are no disynaptic basins (which indicates that the Cs-F bonds are ionic) but you can see two lone pairs on Cs (labels 11 and 12), each basin containing 3.4 electrons. An angle of 164° is subtended at the Cs.
A bit of fun…
Is molecular CsF3 the relevant species here? The literature I have seen proposes CsF3 (with Cs in the +3 oxidation state) as a salt in which the caesium is bonded to only two of the fluorine atoms as CsF2+, the remaining fluorine atoms forming separate fluoride ions. Would CsF2+ avoid the occupation for antibonding orbitals?
D4d-symmetric [CsF8]- is also potentially stable. Bong lengths are less than 2Å there!
Dear Andrey,
Can you elaborate where you got your information from?
An ωB97XD/Def2-TZVPP calculation on the anion CsF8– gives a bond length of 2.01Å, and all positive force constants. The highest, all symmetric Cs-F mode is 493 cm-1. The Cs-F Wiberg bond index is 0.446, and the total Cs bond index is 3.57. There are no significant F-F orders. The HOMO is shown below (antibonding in F-Cs region).
Accordingly, the cation CsF8+ has a shorter Cs-F bond length of 1.916Å, a higher symmetric Cs-F stretch of 538 cm-1, and larger Cs-F Wiberg indices of 0.488 and Cs total bond index of 3.91.
FAIR Data provided at DOI: 10.14469/hpc/10383
Dear Henry,
that was my own computations back in 2016. The idea came from high stability of [XeF8]2- dianion. I even tried to compute the lattice parameters for bcc Cs[CsF8] (space group 89) but ran out of computing resources 🙂
I note that XeF8 has been calculated as being unstable with respect to XeF6 + F2 (DOI:10.1021/ic901956g). It has also been stated that “Rb2XeF8 and Cs2XeF8 are the most stable Xe compounds yet known” but also that “Cs2XeF8 may be considered to be an adduct 2CsF.CsF6“.