Earlier this year, Molnupiravir hit the headlines as a promising antiviral drug. This is now followed by Paxlovid, which is the first small molecule to be aimed by design at the SAR-CoV-2 protein and which is reported as reducing greatly the risk of hospitalization or death when given within three days of symptoms appearing in high risk patients.

The Wikipedia page (first created in 2021) will display a pretty good JSmol 3D model of this; the coordinates being generated automatically on the fly from a SMILES string, which specifies only what atoms are connected in the structure by bonds. Given that the structure of this molecule as embedded in the SARS-CoV-2 main protease[1] has been determined (and can be viewed here), I thought I might display those coordinates as an alternative to the Wikipedia/JSmol generated structure.
Click to get 3D model
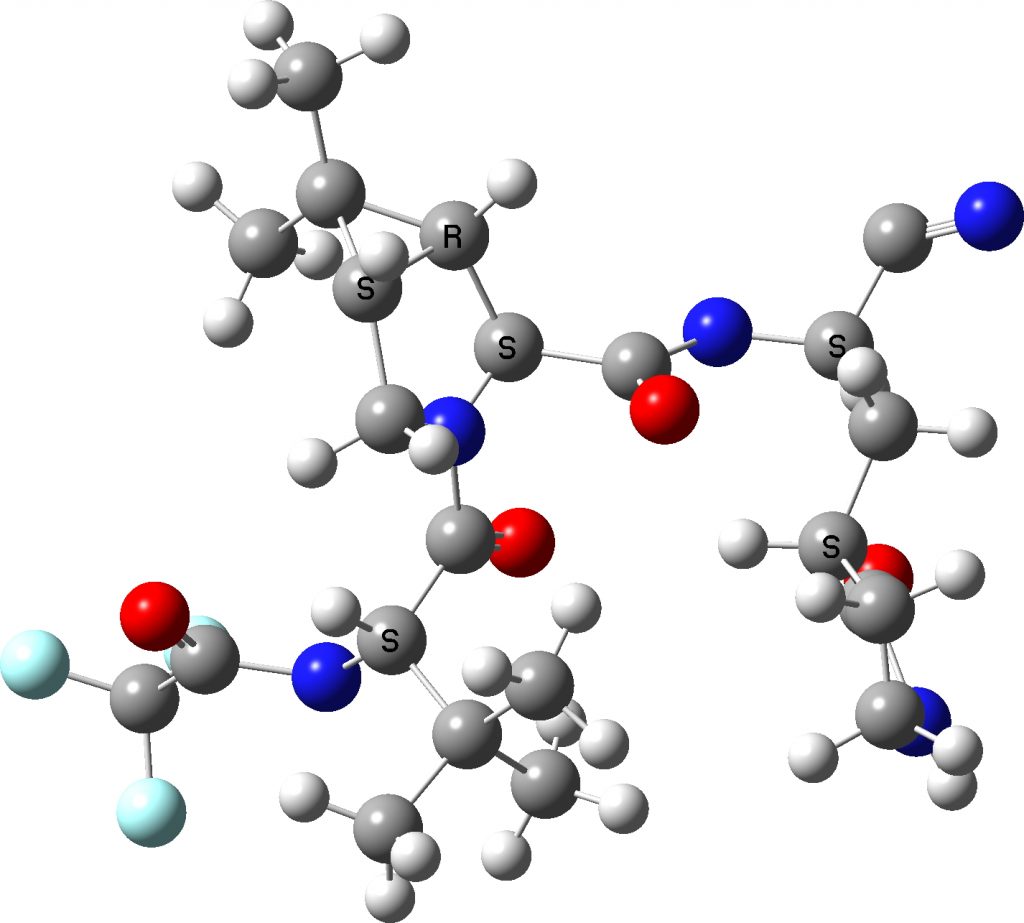
I extracted the ligand from the PDF file and then added hydrogens manually to obtain the above result.‡ There are two noteworthy points about these representations:
- A mystery concerns the nominal C≡N group on the top right, which displays an angle at the carbon of 117°. A cyano group is of course linear (180°). This is not a defect of the crystal structure determination, but an indication of a rather stronger interaction occurring (as indeed noted[1]). The distance between the carbon of the cyano group and an adjacent sulfur is 1.814Å, which indicates a covalent bond has formed to the cyano group. The nitrogen of the erstwhile cyano group is 3.013Å away from an adjacent NH group, which suggests it is stabilised by a hydrogen bond.
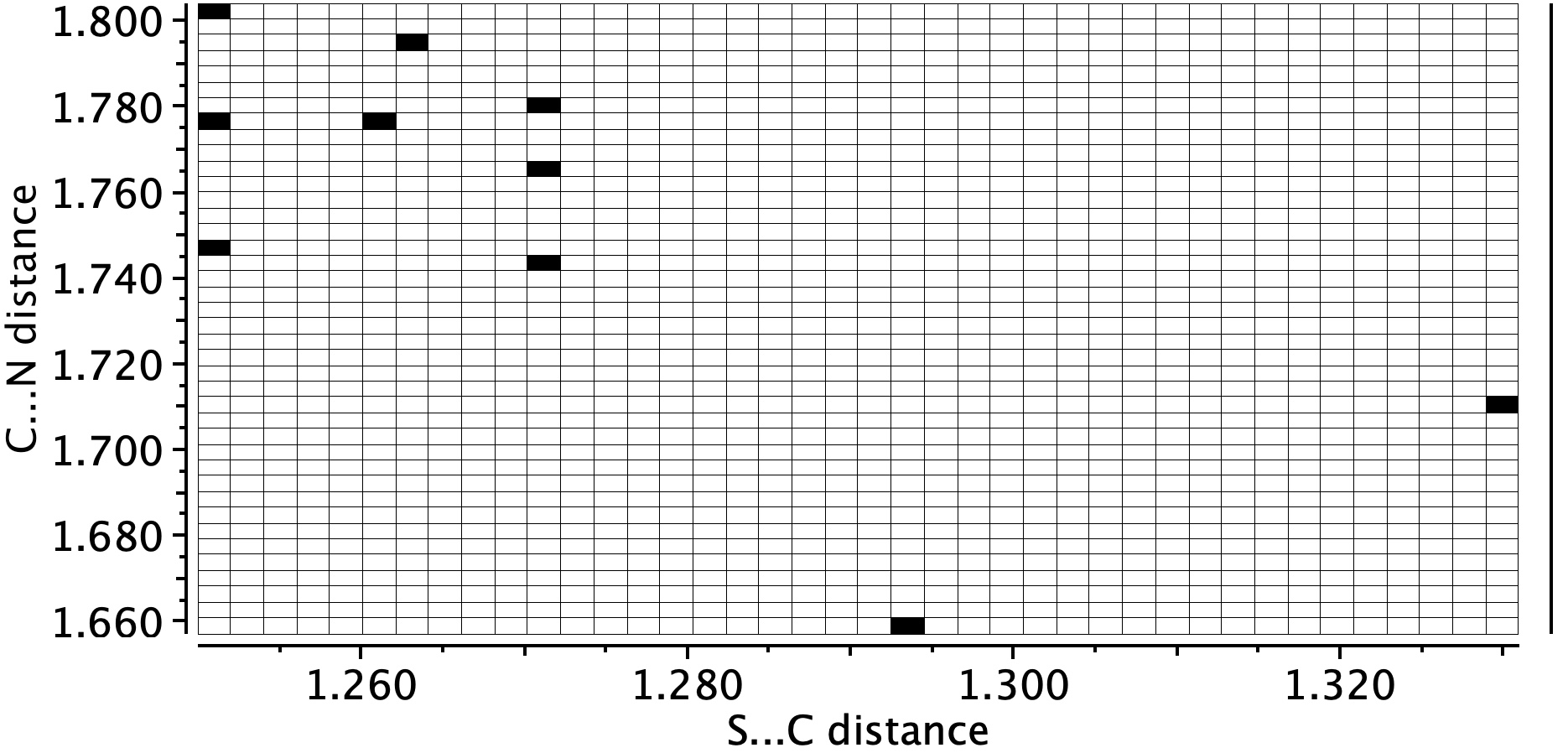
- Crystal structure searching of units with S…C…N in which the N has only one bond reveals zero hits, but searches of S…C…NH reveal nine hits, with S…C distances in the range 1.74 – 1.80Å and C…N distances in the region 1.25-1.27Å. The reported CN distance is 1.251&ARing, confirming that when bound to the protein, the cyano group is replaced by an S-C=NH group and hence is clearly an important component of the mode of action of Paxlovid.
- The conformation of Paxlovid is in one respect not fully represented by the Wikipedia diagram, as shown below. This implies the t-butyl group (on the left) as being well separated from the pyrrolidinone ring system at the right of the molecule.

In fact the two groups are adjacent, being held in that conformation by probably a combination of weak dispersion forces and a contribution from the surrounding protein in the crystal structure. This is more graphically shown by the NCI (non-covalent-interaction) diagram below (DOI: 10.14469/hpc/9964), where the green areas in the region between the two groups (ringed in red) represent stabilising interactions between them. You might also spot other green/cyan regions indicating additional weak hydrogen bonds between C-H groups and oxygen!
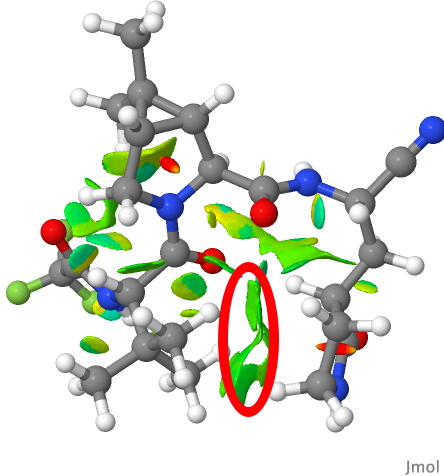
PAXLOVID NCI analysis
There are only a small number of crystal structures of small molecules containing the S-C=NH motif. I will try to find out how common this is in protein-ligand structures.
‡ There are many tools for performing this operation. I used the following procedure. I downloaded the PDB file (https://files.rcsb.org/download/7vh8.cif), opened it in CSD Mercury, selected the ligand (by identifying the CF3 group and clicking on one atom), inverted the selection so that everything but the ligand was then selected and using edit/structure, I deleted the selected atoms, leaving only the ligand.
Postsript
The cyanopyrrolidine group such as in Paxlovid is well known as a specific probe.[2],[3],[4] CovalentInDB is a comprehensive database facilitating the discovery of such covalent inhibitors[5] and is available here. There is also a program called DataWarrior that is potentially able to find such probes.
Author
References
- Y. Zhao, C. Fang, Q. Zhang, R. Zhang, X. Zhao, Y. Duan, H. Wang, Y. Zhu, L. Feng, J. Zhao, M. Shao, X. Yang, L. Zhang, C. Peng, K. Yang, D. Ma, Z. Rao, and H. Yang, "Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332", Protein & Cell, vol. 13, pp. 689-693, 2021. https://doi.org/10.1007/s13238-021-00883-2
- N. Panyain, A. Godinat, A.R. Thawani, S. Lachiondo-Ortega, K. Mason, S. Elkhalifa, L.M. Smith, J.A. Harrigan, and E.W. Tate, "Activity-based protein profiling reveals deubiquitinase and aldehyde dehydrogenase targets of a cyanopyrrolidine probe", RSC Medicinal Chemistry, vol. 12, pp. 1935-1943, 2021. https://doi.org/10.1039/d1md00218j
- N. Panyain, A. Godinat, T. Lanyon-Hogg, S. Lachiondo-Ortega, E.J. Will, C. Soudy, M. Mondal, K. Mason, S. Elkhalifa, L.M. Smith, J.A. Harrigan, and E.W. Tate, "Discovery of a Potent and Selective Covalent Inhibitor and Activity-Based Probe for the Deubiquitylating Enzyme UCHL1, with Antifibrotic Activity", Journal of the American Chemical Society, vol. 142, pp. 12020-12026, 2020. https://doi.org/10.1021/jacs.0c04527
- C. Bashore, P. Jaishankar, N.J. Skelton, J. Fuhrmann, B.R. Hearn, P.S. Liu, A.R. Renslo, and E.C. Dueber, "Cyanopyrrolidine Inhibitors of Ubiquitin Specific Protease 7 Mediate Desulfhydration of the Active-Site Cysteine", ACS Chemical Biology, vol. 15, pp. 1392-1400, 2020. https://doi.org/10.1021/acschembio.0c00031
- H. Du, J. Gao, G. Weng, J. Ding, X. Chai, J. Pang, Y. Kang, D. Li, D. Cao, and T. Hou, "CovalentInDB: a comprehensive database facilitating the discovery of covalent inhibitors", Nucleic Acids Research, vol. 49, pp. D1122-D1129, 2020. https://doi.org/10.1093/nar/gkaa876
Nice web page Henry! One small thing: the paxlovid jsmol structure has a “C=N” where there should be a nitrile group. The bond angle is about 120 deg rather than required 180 deg.
John,
The odd angle at the CN group is from the coordinates from SARS-CoV-2 main protease and appears not to be an artefact. I discuss this feature above. Jsmol determines the bond order from the length, and since it is 1.25A, it has selected a double bond. Remember, almost all bond orders displayed in such structures are not real but are determined from the bond lengths! Indeed, if it were to be a triple bond, the bond angle would be very anomalous.