
Scheme 1

Scheme 1
Of the four heterocyclic ring systems 1a-d that we chose to investigate, variously substituted examples of all but the last have been characterised crystallographically.5 These structures reveal that only a tub or boat shaped conformation is known, referred to here as having Cs symmetry (when symmetrically substituted). As planar species, such 4n-p electron rings would be formally anti-aromatic, and distortion from C2v to Cs symmetry, with the accompanying bond localisation, is the normal route for reducing or eliminating anti-aromaticity. A corresponding system having some Möbius character, and with only a C2 axis of symmetry, is another, hitherto unexplored alternative, with the potential for reversing the anti-aromaticity into aromaticity. We chose two strategies to investigate whether such conformations might be energetically accessible. The first involved perfluorination, which we have previously suggested6 has the effect of reducing twist alternation in such rings and increasing aromaticity. Our second strategy was to promote C2 symmetry and suppress Cs symmetry by constructing the 7-membered ring from the basic biphenyl motif 3. We also included the bicyclic forms 2, which are known to be in equilibrium with 1, for comparison.
Considering firstly the perfluoro-oxepine 1a, vibrational analysis of the planar system (of C2v symmetry) predicts a single imaginary mode (Table) corresponding to a distortion to the lower Cs symmetry. Geometry optimization starting from a high twist C3-C4-C5-C6 dihedral angle results in cis to trans isomerism of the central formal double bond and location of a minimum with C2 symmetry. This isomer, although 21.8 kcal mol-1 higher in energy than the planar C2v saddle point, has a NICS value suggesting some significant aromaticity. The presence of one pseudo-trans component was characteristic of the C9H9+ species identified by Schleyer2 and has also been recently noted for cyclo-octatetraene.11 The ring bond lengths of C2-symmetric cis,trans,cis 1a also reveal reduced alternation, characteristic of increased aromaticity or decreased anti-aromaticity (Figure 1).
Two other isomers can be found for 1a. The first results from distortion from the C2v saddle point to the "tub" mode typical of 4n p-electron all-cis cyclo-octatetraene and corresponds to the conformation observed in the crystal structures4a. The second isomer, of very slightly lower energy, is the perfluorinated analogue 2a of the known benzene oxide 2. The C-C bond lengths of the C2-symmetric cis,trans,cis isomer of thiepine 1b indicate a slightly reduced tendency toward aromaticity for this species, compared to the oxepine (Figure 1).
With the nitrogen analogue, the C2v geometry
reveals a bond length pattern (Figure 2) at the planar geometry
which corresponds to the specific valence bond isomer 5c
(Scheme 2) one of the two possible forms resulting from a
Jahn-Teller like distortion of the formally anti-aromatic 4n
ring.

Scheme 2
A high positive NICS value (17.5) confirms significant anti-aromaticity (the high value may also be due to some contamination from a smaller HOMO-LUMO energy gap). Further distortion to alleviate this anti-aromaticity is implied by the existence of two imaginary normal modes deriving from the Hessian matrix (i.e. this geometry is a second-order saddle point), each corresponding to Cs distortion to a lower energy form corresponding to that of the crystal structure.4a
Like the oxygen and sulfur systems, a C2 symmetric form can be located which reveals an inverted NICS value indicative of moderate aromaticity. This species differs from the oxa and thia counterparts in two important ways. Firstly, the energy is lower rather than higher than at the C2v geometry (by 9.2 kcal mol-1). Secondly, no trans ring component is present, and the bond length pattern is different, corresponding more closely to that for the chiral species 7c (Figure 2).
Both these differences require more detailed analysis. The lower energy appears to contradict the form of the two imaginary normal modes noted above for 5c, for which there is no C2 symmetric distortion resulting in an energy decrease. The contradiction can be rationalised by inspecting an orbital correlation diagram between the C2v (5c) and C2 (7c) geometries (Figure 3). This shows 5c to have four occupied p orbitals, three of b1 and one of a2 symmetry. The b1 HOMO corresponds to valence bond isomer 5c (and the LUMO to the other possible isomer 6c). However, 7c has two occupied orbitals deriving from the pp orbitals of b and two of a form. This means that the planar (anti-aromatic) Hückel isomer 5c correlates with the doubly excited state of chiral 7c, and vice versa. Such a result has indeed been previously observed for the related system 1,2,4,6-cycloheptatetraene,6,12 for which chiral inversion as a closed shell species is forbidden by orbital symmetry to pass through a planar form and must instead proceed through an open shell singlet state transition state.
The analogy to cycloheptatetraene also extends to the bond length pattern of 7c, for which we note the unusually long predicted N-F bond length (Figure 2). Before discussing this aspect in further depth in the next section, we note for completeness at this stage a further cis,trans,cis ring isomer of 7c which can also be located. Rather than having C2 symmetry, the nitrogen-fluorine axis is distorted away from the C2 axis. This avoids destabilising repulsions between the occupied p orbital of the remote ring trans double bond and that of the s N-F bonding orbital. This isomer is calculated to lie 9.8 kcal mol-1 above the planar stationary point 5c, and in this relative energy and also the bond length pattern (Figure 2), more closely resembles the results for the oxepine than for 7c.The remarkably long N-F bond length (1.688Å) for 7c compared to the value for 5c at the planar geometry (1.374Å) or indeed for the cis,trans,cis isomer (Figure 2) requires an explanation. Firstly, we established that this value depends critically on the nature of the two atoms comprising this "single" bond. Replacing N-F with the isoelectronic C--F gives a normal value of 1.379Å but equally elongated values are calculated for the other halogens (C2/C2v): N-Cl 2.463/1.719, N-Br 2.568/1.880, N-I 2.794/2.092 (at the extended B3LYP//6-311G(d) basis set level). This suggests the origins of the effect are related to the relatively low lying energy of the N-Halogen s* anti-bonding orbital, and its resulting interaction with high energy ocucpied orbitals of the same ai symmetry. The effect is also dependent on the correlation treatment. At the Hartree-Fock level, the N-F bond length is almost normal (1.39Å), but at correlated levels it is significantly lengthened (R3BYLP; 1.69, MP2; 1.70). The unusual nature of the N-halogen bond located on the C2 axis in these systems therefore arises at least in part from a correlated interaction of the electrons in this region.
To probe this aspect further, we inspected the characteristics of the highest occupied orbital of a symmetry in 7c. Qualitatively, this occupied orbital originates from the mixing induced by twisting the anti-bonding p orbital of a2 symmetry and the anti-bonding s orbital of a1 symmetry in 5c. Such mixing is of course allowed in C2 symmetry. The effect is that one phase of the forming orbital passes directly through the C2 axis coincident with the N-F bond, in the process establishing an anti-bonding node along this bond (Figure 3). In this simple interpretation, the cumulative effect of creating a Möbius phase inversion by twisting the pp system is to lengthen the N-F bond by 0.313Å.
As we noted above, such mixing requires orbital occupancy corresponding to the doubly excited state of 5c. The optimised planar geometry of this state (Figure 2) corresponds closely to the representation 8c, and its energy is 13.7 kcal mol-1 above that of 5c. The vibrational analysis, unlike that of 5c however, now does show an imaginary mode of C2 symmetry, implying the potential energy surface is continuously connected to 7c. This is verified by inspection of the orbital correlation (Figure 3), which reveals how the three p and one s orbitals of 8c (the latter located in the N-F region) do correlate into the top four occupied orbitals of Möbius p character for 7c. Other orbitals shown in the diagram include the HOMO of 8c (a fluorine lone pair of b1 symmetry) and the a1 symmetric N-F s bonding orbital, both of which correlate with lower lying b orbitals in 7c. The overall correlation in fact is strikingly similar to that previously presented for 1,2,4,6-cycloheptatetraene and cycloheptatrienylidene carbene6. To employ a more familiar metaphor, the N-F bond in this system could be described as a screw-on/screw-off type, since its length depends on the degree of twist!
Analogue 1d shows behaviour different in several regards to that of nitrogen. A stationary point with C2v symmetry was initially located with a bond length pattern corresponding to 6d (Figure 4). The Hessian reveals it as a third order saddle point with three imaginary normal modes, the displacement vectors for first two corresponding to C2, and third to Cs non-planar distortion. Appropriately, 6d has two p orbitals of b1 and two of a2 symmetry, and so now correlates with the ground (rather than the excited) state of the Möbius isomer 7d (Figure 5), which also rationalises the presence of C2 distortive normal modes. The NICS values again show a large inversion from anti-aromaticity to aromaticity. The difference between N and P lies in the nature of the heteroatom. With the more electronegative N, the highest occupied orbital (HOMO) has density (i.e. no node) on the heteroatom, whereas the less electronegative phosphorus shows no density at the HOMO.

We anticipated that, unlike the azo-analogue, a relaxed potential scan (Figure 6) starting from the 7d structure and incrementally reducing the C2 twist would continuously connect to 6d. However, the energy of the planar resulting species was 14.9 kcal/mol lower than 6d, and with a structure corresponding approximately to 8d. Non-alternating ring bond lengths and the aromatic NICS value (-7.9 ppm) derive from some phosphatropylium fluoride ion pair character for 8d, but the P-F bond length is much less extended than the nitrogen equivalent. An orbital correlation diagram (Figure 6) reveals that the process of converting 7d to 8d again correlates only six of the eight ring p electrons, the remaining two converting to s electrons occupying the P-F region, but this time in a manner which is significantly less anti-bonding along the P-F axis. Similarly, 7d exhibits an almost normal P-F bond length, reflecting the greater degree of five-valency possible for phosphorus via d-orbital participation. The reduced ionic character appears associated with a greater NICS aromatic value for 7d.
Solvation has little effect on the relative energies (Table); neither 7d nor 8d is significantly stabilised with respect to the lowest energy species of Cs symmetry, again confirming the higher hypervalency of the phosphorus centre. In contrast, the Möbius form 7c shows extension of the N-F bond to 2.03Å but the solvation free energy only decreases about 5 kcal mol-1 compared to the Cs conformation. The fully dipolar isomer 8c reveals a decrease of about 24 kcal mol-1, confirming its greater ionic characteristics. The effect of such solvation is to decrease the predicted chiral inversion barrier for 7c.
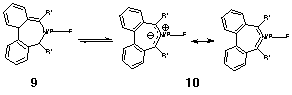
To see if we could design a system where this energy difference might be low enough to enable experimental detection of Möbius conformations, we investigated the dibenzo derivatives. Here, the tendency to retain aromaticity in the benzo ring was designed to inhibit the Cs symmetric form, and this is indeed verified in the calculations, where no such stationary point could be located. However, ring closure to give 4 resulted in a lower energy species than the C2 isomer 3 which were between 40-53 kcal/mol higher in energy. These isomers also displayed another significant difference from the monocyclic forms. The NICS values at the 7-ring centroid indicate significant anti-aromaticity and the 6-rings are surprisingly non-aromatic. The HOMO for the aza system 3/10 (Figure 7), although C2 symmetric, is quantitatively different from that for 7c (Figure 3) in having little density at the ring centroid. Replacing R=H by R=CN (which promotes the dipolar resonance form 10) enhances the Möbius characteristics, but insufficiently to render this the most stable form.
| Table 1. Energies, kcal mol-1, relative to 2 or 4 (B3LYP/6-31G(d) in Hartree) and computed NICS Values (ppm). | ||||||
|---|---|---|---|---|---|---|
| System | Point Group | C=C configuration |
Negative Roots |
Dihedral angle C3-C4-C5-C6 |
Energy relative to 2 or 4 (total energy in parentheses) |
NICS(0) |
| 1a | Cs | cis,cis,cis | 0 | 0 | 2.6 (-902.7656) | -5.7 |
| 1a | C2 | cis,trans,cis | 0 | 94.8 | 36.3 | -10.5 |
| 1a | C2v | cis,cis,cis | 1a | 0.0 | 14.5 | +9.4 |
| 1b | Cs | cis,cis,cis | 0 | 0 | 4.7 (-1225.7418) | -8.0 |
| 1b | C2 | cis,trans,cis | 0 | 107.7 | 36.7 | -10.9 |
| 1b | C2v | cis,cis,cis | 1b | 0.0 | 21.3 | +7.3 |
| 1c | Cs | cis,cis,cis | 0 | 0 | -21.3 {-24.7}g (-982.0395) | -9.5 |
| 1c | C2 | cis,cis,cis (7c) | 0 | 32.4 | 4.9 {-1.1}g | -6.6 |
| 1c | C1 | cis,trans,cis | 0 | 98.1 | 23.9 | -10.7 |
| 1c | C2v | cis,cis,cis (5c) | 2c | 0.0 | 14.1 | 17.5 |
| 1c | C2v | cis,cis,cis (8c) | 2d | 0.0 | 27.8 {4.2}g | - |
| 1d | Cs | cis,cis,cis | 0 | 0 | -11.4 {-9.8}g (-1268.7453) | -7.9 |
| 1d | C2 | cis,cis,cis (7d) | 0 | 43.9 | 46.0 {47.7}g | -10.9 |
| 1d | C1 | cis,trans,cis | 0 | 111.0 | 23.0 | -6.5 |
| 1d | C2v | cis,cis,cis (6d) | 3e | 0.0 | 81.9 | 53.4 |
| 1d | C2v | cis,cis,cis (8d) | 3f | 0.0 | 67.0 {63.1}g | -8.5 |
| 3a | C2 | cis,cis,cis | 0 | 39.4 | 40.5 (-614.7114) | 26.5, -1.1h |
| 3b | C2 | cis,cis,cis | 0 | 42.7 | 25.5 (-799.1745) | 16.5, -3.1h |
| 3c | C2 | cis,cis,cis | 0 | 53.8 | 46.2 (-937.7039) | 14.9, -4.0h |
| 3d | C2 | cis,cis,cis | 0 | 41.2 | 19.3 (-594.8438) | 20.8,-1.0h |
| 3e | C2 | cis,cis,cis | 0 | 58.4 | 52.6 (-881.4595) | 8.2, -6.2h |
| 3f | C2 | cis,cis,cis | 0 | 60.2 | 34.6 (-1065.9222) | 4.0, -7.7h |


| 5c | Symmetry | 7c | 8c | Symmetry | |
|---|---|---|---|---|---|
 |
a2 | a |  |
 |
b1 |
 |
b1 | a |  |
 |
a2 |
 |
a1 | b |  |
 |
a1 |
 |
a1 | a |  |
 |
a2 |
 |
a2 | b |  |
 |
b1 |
| HOMO-LUMO gap | |||||
 |
b1 | a |  |
 |
b1 |
 |
a2 | a |  |
 |
a1 |
 |
b1 | b |  |
 |
a2 |
 |
b1 | b |  |
 |
a1 |
 |
b1 | b |  |
 |
b1 |
 |
a1 | a |  |
 |
b1 |

| 5d | Symmetry | 6d | 7d | Symmetry | |
|---|---|---|---|---|---|
 |
a2 | b |  |
 |
a2 |
 |
b1 | a |  |
 |
b1 |
 |
b1 | b |  |
 |
b1 |
| HOMO-LUMO gap | |||||
 |
a2 | a |  |
 |
a1 |
 |
a2 | a |  |
 |
b1 |
 |
b1 | b |  |
 |
a2 |
 |
b1 | b |  |
 |
b1 |
 |
b1 | b |  |
 |
b1 |

