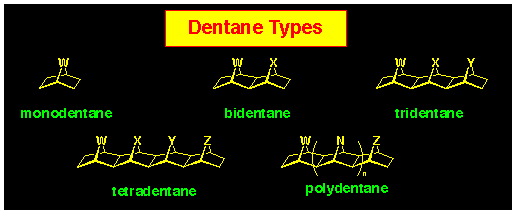
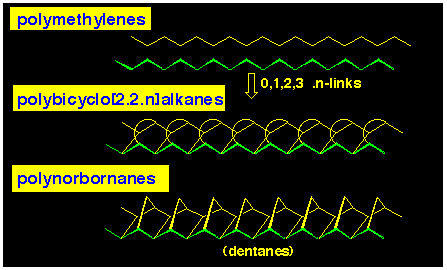
We have coined the 'dentane' name for the exo, exo-fused
polynorbornane series of compounds in which the bridges have a
syn-facial relationship (Figure 4). It derived this generic
name as an extension of the [3]polynorbornane system where the
3bridges were likened to a trident (see next subsection).
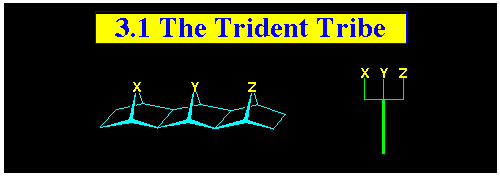
3.1 Tridents, Especially Those with
a Central Nbridge (Y = NR)
The trident series is a subsection of the dentane family (see
Section 3) of polynorbornanes containing 3-fused norbornanes.
They have been so named because the three syn-facial bridges
in the [3]polynorbornanes are structurally similar to the top
section of a trident.
We have used the trident system as our testing ground for the
ACE and aza-ACE methodology in order to assess coupling selectivities.
In particular, we have determined the compatibility of juxtaposed
heterobridges in these systems with a view of applying this knowledge
to larger systems (arc-shaped molecules, polarofacials, Section
3.2, 3.3 respectively).
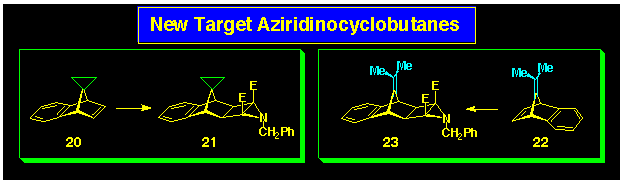
The stereoselectivity of the aza-ACE coupling reaction has been
evaluated using the aziridinocyclobutane 7 as the 4p-reagent
and various benzonorbornadienes as the 2p-reagents,
following on the early work with norbornadiene (Section 2, Scheme
3).
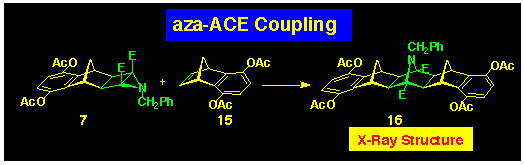
A key finding was provided by the reaction of the diacetoxybenzonorbornadiene
15 with aziridine 7 which produced the symmetrically
coupled product 16 (Scheme 5). The 1H
NMR spectrum of this product was fully symmetrical at room temperature
and did not change on heating. This result could be interpreted
in a number of ways.
1) The nitrogen substituent is sp2 hybridised and switching very rapidly from one side of the molecule to the other in a degenerate interconversion.
2) The bridge nitrogen is sp2 hybridised.
3) The bridge nitrogen is somewhere in between sp2 and sp3.
In fact, the X-ray structure of 16 indicates that explanation
3 is correct, at least in the solid state. We are currently investigating
this feature by 15N NMR spectroscopy as part
of a wider investigation of the effect of flanking substituents
on the structure of the central N-bridge in the XNY-trident series.
In the first stage of this work, we have been able to show that
the aza-ACE coupling reaction is extremely tolerant of the nature
of the bridging atom in the 7-position of the benzonorbornadiene
and yields trident systems with both the 7-oxa and 7-aza-benzonorbornadienes
(Scheme 6).

We are presently preparing the NNX-tridents and ONX-trident systems
(Scheme 6, X= NR and O respectively) as additional models for
this study. In addition, we are progressing the program by introducing
carbon substituents into the 7-position as part of this study
(Scheme 7).
3.2 Polarofacial Poly(Heterobridged-Norbornanes)
When we first considered the concept of polynorbornanes formed
from the assembly of 7oxanorbornanes, we naively thought
that the alicyclic framework would be straight. Accordingly,
we envisaged that they could act as ionophores for transport of
ions across membranes. This false impression was gained by considering
their formation as being derived from two polymethylene chains
and linking these alternatively with zero and methylene bridges (see Figure 5).
However, molecular modelling showed that this was not the case,
and that we are dealing with curved structures.
3.2.1 The Arc-shaped Framework
The concept that polynorbornanes were in fact curved rather than
rod-like, had a large bearing on the way we approached the design
of rigid molecular architectures. Indeed the positioning of chromophores
at the termini of molrac systems composed of polynorbornanes meant
that the relative orientation of such effector groups would be
changed depending on the geometry of the frame. Indeed, this
could be used to aid design and fine tune effector orientation,
based on a knowledge of the curvature of the frame. This led
to the concept of arc-shaped topography (Figure 6), where the
curvature of the arc was governed by the nature of the bridges
in the component norbornane subunits.
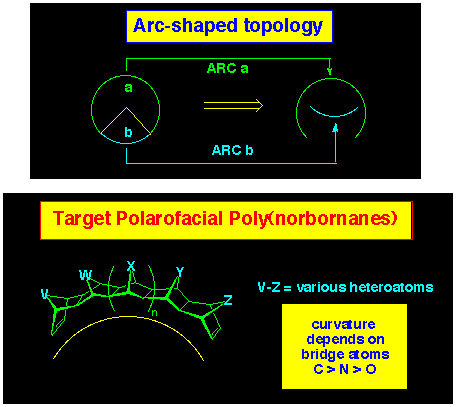
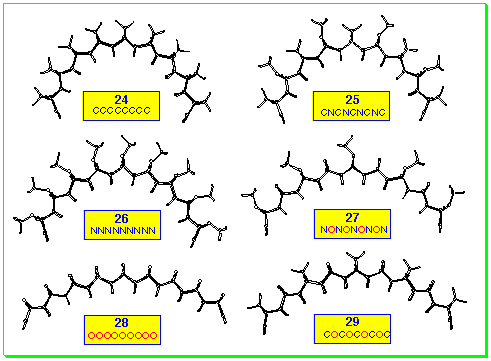
The curvature of the arc-shape systems was evaluated using molecular
modelling conducted at the AM1 level and a selection of polynorbornane
systems are displayed in Figure 7.
It is immediately apparent that the radius of curvature of the
polynorbornanes differ significantly: the all-carbon system being
the most curved while the all-oxygen system has the least curvature.
It is of practical significance that the N-bridged systems
are almost as curved as the carbon systems, since the former have
not been described with more than two fused norbornanes, whereas
the all-nitrogen systems are accessible through aza-ACE coupling.
Mixed systems of CN, CO, NO, and CNO have all been prepared and
judicious choice of bridge partners allows tuning of the curvature
in the polynorbornane system.
3.2.2 The Poly(7oxanorbornanes)
In earlier approaches to the synthesis of poly(7-oxanorbornanes)
we had explored the reaction of isobenzofurans with 7-oxanorbornenes
and found that both exo,exo-isomers and exo,endo-isomers
were produced. The stereoselectivity of these cycloadditions
could be influenced by modification of the bridgehead position
of the isobenzofuran, and we exploited this in the preparation
of bis(crown ethers). While it was appropriate to use
unsubstituted crown-isobenzofurans for the preparation of cavity
shaped bis(crown ethers) (see Section 4, Scheme 29), formation
of extended frame products required the use of arylated isobenzofurans.
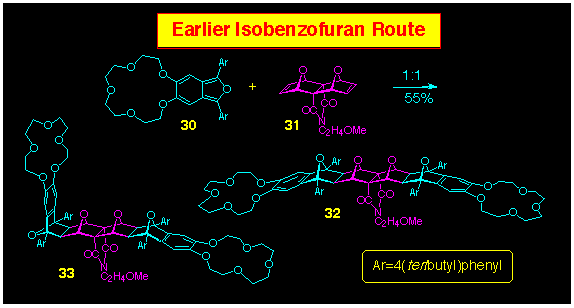
Thus, reaction of the bisalkene 31 with arylated
isobenzofuran 30 provided access to the extended frame
product 32 with four contiguous O-bridges, together
with the half-bent isomer 33 (Scheme 8). Such isobenzofuran
reagents were inappropriate, however, for the production of more
extended bis-crowns until appropriate bis(7-oxanorbornenes)
were available. Work in this area has been largely overtaken
by the successes of the BLOCK protocols described
later (Section 4).
The opportunity to use BLOCK coupling techniques
in this work appeared promising when the ACE coupling reaction
was found to produce exclusively extended-frame products on reaction
cyclobutene epoxides with norbornenes (vide supra, Scheme
1). We were hopeful that similar stereoselectivity would be obtained
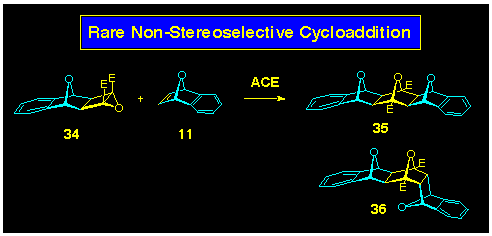
with 7oxanorbornenes, however, this was not to be, and both
stereoisomers 35 and 36 were produced in the reaction
of 7-oxabenzonorbornadiene 11 and the cyclobutene epoxide
34 (Scheme 9). It is interesting to note that the adverse
oxygen, oxygen orbital interactions which contribute to the production
of the bent-frame isomers such as 36 are apparently not
as strong in nitrogen, oxygen or nitrogen, nitrogen systems (currently
under theoretical study). Accordingly, reaction of 7-azabenzonorbornadiene
with cyclobutene epoxides again exhibits extended-frame stereoselectivity
(vide supra).
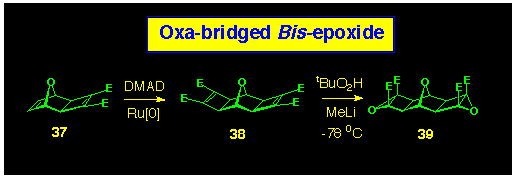
Notwithstanding the lack of stereoselectivity of the ACE reaction
with 7-oxanorbornenes, we pressed on to make the oxygen-bridged
bis-epoxide reagent 39 (Scheme 10). This was achieved
by reaction of the previously reported bis-alkene 37
with DMAD in the presence of a ruthenium catalyst to produce
the bis(cyclobutene-1,2-diester) 38, which was epoxidised
(tBuO2H,MeLi, -78 oC)
to form the bis-epoxide 39.
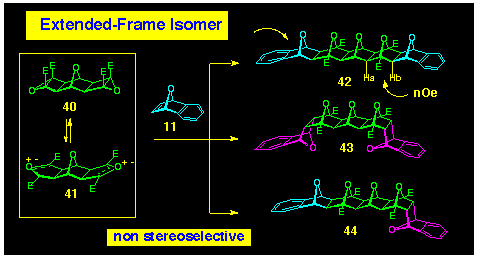
The reaction of 7-oxabenzonorbornadiene 11 with the bis-epoxide
39 was found to produce three isomeric cycloaddition compounds
42-44 in a 1:1:2 ratio (Scheme 11). The major product
44 is readily identified (four singlet CH resonances, three
oxa-bridgehead resonances and two ester methyl resonances), however,
the C2v symmetry of the other products 42,
43 renders their spectra both simple and similar. The
distinction cannot be made reliably on chemical shift data alone
and was achieved using nOe data. The required polarofacial system
42 shows a clear nOe between protons Ha and Hb on the lower
face of the molecule, thereby clinching its stereochemistry.
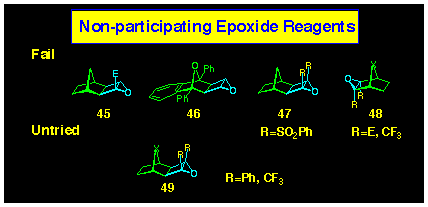
The ACE coupling is very specific regarding the nature of the
epoxide which will participate in the reaction. To date it is
restricted to cyclobutene epoxides containing two ester activating
groups and simple changes, eg removal of one ester 45,
both esters 46 or their replacement with phenyl sulfonyl
groups 47, negates their participation (Figure 8). Further,
norbornene epoxides 48 do not participate and can
be incorporated into the ACE reagent with immunity. We have exploited
this fact to increase the length of the oxygen substituted face
of these polarofacials.
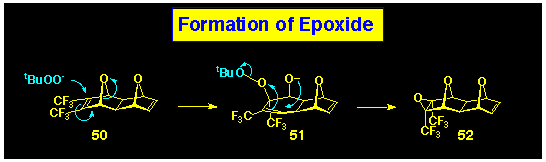
The required ACE BLOCKs containing the non-participating
norbornene epoxide are each prepared from the 2:1-adduct 50,
obtained from reaction of furan with perfluorobut-2-yne. It is
interesting to note that the nucleophilic epoxidation conditions
used to convert the cyclobutene-1,2-diester to their epoxides,
also effects epoxidation of the trifluoromethyl-substituted pbond
in 50. This does not occur in related norbornene systems
and clearly involves participation of the oxygen bridge. A mechanism
for this transformation is outlined in Scheme 12, and finds recent
precedent in the attack of carbanions onto 7-oxanorbornenes.ref
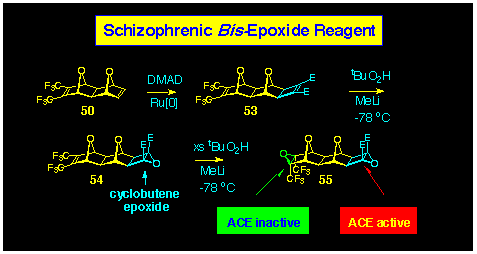
Ruthenium-catalysed addition of DMAD to the 7-oxanorbornene 50
occurs site selectively at the unsubstituted alkene to yield the
cyclobutene-1,2-diester 53 (Scheme 13). Treatment with
tertiary butyl peroxide under the normal low temperature conditions
causes epoxidation to occur at both p-centres
of 53 to form bis-epoxide 55; controlled
epoxidation allows isolation of the mono-epoxide 54, showing
there is a clear preference for attack at the cyclobutene p-centre.
Having ACE BLOCKs of each type
with inbuilt epoxides has allowed the preparation of several new
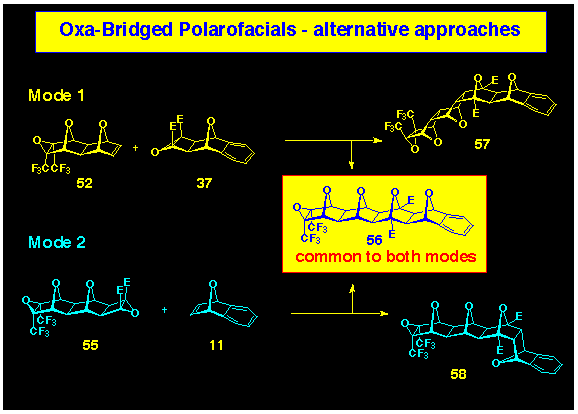
polarofacial systems. Compound 56, with five juxtaposed
oxygen bridges, was prepared in two ways, one where the epoxide
was provided by the alkene BLOCK (Scheme
14, mode 1 ) and the other where it was an end component of the
cyclobutene epoxide BLOCK 55 (Scheme
14, mode 2). In each coupling mode, the required polarofacial
target 56 was accompanied by roughly equal amounts of an
isomer, the structure of which depended on the mode of coupling,
eg 57 from mode 1 and 58 from mode 2 (Scheme 14).
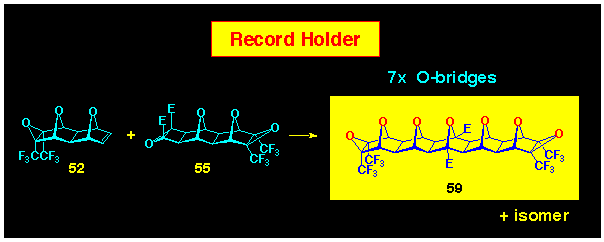
The coupling of the ACE BLOCKs 52 and
55 which each carry the inert epoxide functionality can
also be achieved and this leads to the seven oxygen-bridged system
59; again it was accompanied by its stereoisomeric shadow
(Scheme 15). This is the most extended
oxa-bridged polarofacial system ever made, although
the methodology does allow the potential to form more extended
systems.
3.2.3 The Aza-bridged
Polarofacial systems
The major advantage of working in the nitrogen-bridged series
is that the coupling reactions are all stereoselective and, as
a consequence, single products are formed. As presented earlier
(Scheme 3), the aza-ACE reaction proceeds
with exo,exo-stereochemistry to afford extended-frame products.
On the experimental side, the aza-ACE
reaction occurs at a significantly lower temperature (80 oC)
than the ACE reaction (140-160 oC)
and this means that the reaction can be achieved by reflux in
benzene without recourse to sealed tube conditions.
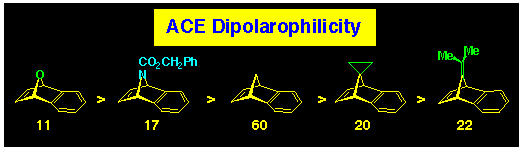
We find that the 7-substituent of the benzonorbornadiene effects
the rate of reaction (dipolarophilicity) towards aza-bridged aziridines
in the aza-ACE reaction. 7Oxa-benzonorbornadiene 11
is the most reactive, while 7-isopropylidene-benzonorbornadiene
22 is the least reactive; other substituted members are
of intermediate reactivity (see Scheme 16).
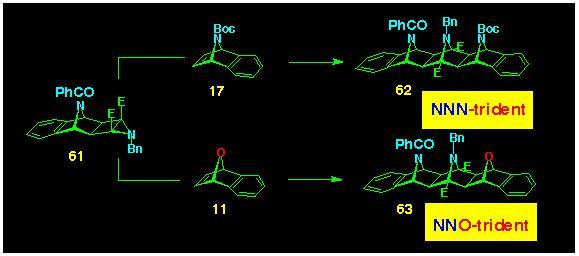
The basis for the entry to all-heterobridged systems was provided
by N-bridged aziridine BLOCK 61
which yielded the NNN-trident 62 on reaction with the N-bridged
benzonorbornadiene 17 and the NNOtrident 63
by coupling with 7-oxabenzonorbornadiene 11 (Scheme 17).
Again, the 1H NMR spectra of these products
are complicated by loss of symmetry associated with N-substituent
mobility at room temperature and only return to C2v
or Cs-symmetry at higher temperature.
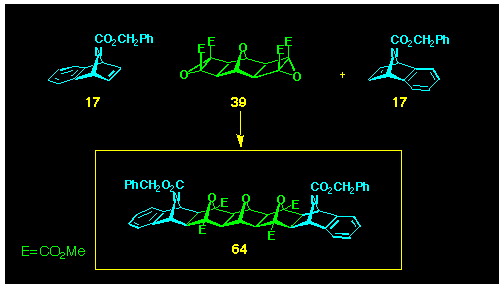
The larger mixed oxygen, nitrogen systems have been produced
using ACE coupling on dual epoxide BLOCKs
and N-bridged benzonorbornadienes. Thus, reaction of bis-epoxide
40 with NBoc-7-azabenzonorbornadiene 17
(140 oC, DCM) produced the pentadentane 64
(Scheme 18).
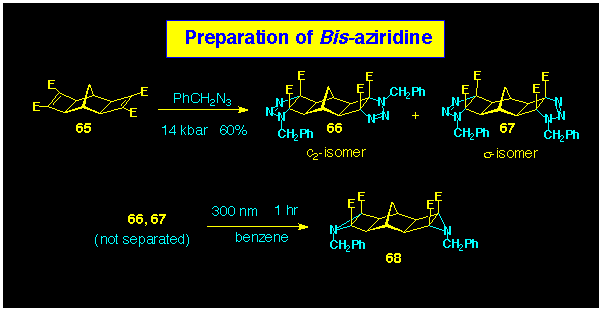
For the study of multiple N-bridged dentane structures,
we have again drawn on the dual coupling protocol. In particular,
we have prepared the bis-aziridine 68, using the
method outlined in Scheme 19. In this case, addition of azide
could only be achieved using high-pressure conditions and then,
with only moderate efficiency. A mixture of C2
and s-isomeric bis-triazolines
66 and 67 were produced but these were not separated
as each yielded the required dual BLOCK 68
upon irradiation.
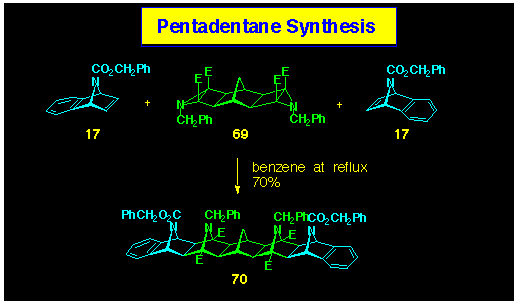
Reaction of the N-bridged benzonorbornadiene 17
with dual aziridine 68 was conducted in benzene solution
at reflux and produced the [5]polynorbornane 70 in which
four of the syn-facial bridges are nitrogen (Scheme 20).
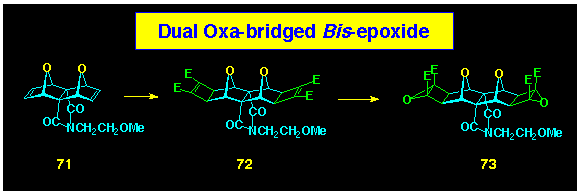
The synthesis of the dual oxygen-bridged BLOCK
73 in the cyclobutene epoxide series was prepared in two
steps from the known bis-alkene 71 according to
our standard protocol (Scheme 21). This complements the single
oxygen bridged system 39 described earlier.
To date, we have not been successful in obtaining the bis-aziridines
corresponding to epoxides 39 and 43 as addition
of benzylazide fails to add onto the oxa-bridged cyclobutene-1,2-diesters
38 and 72. We are presently investigating alternative
ways of converting cyclobutene-1,2-diesters to aziridinocyclobutanes.
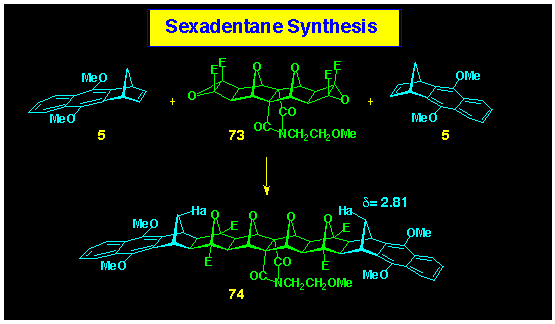
The preparation of sexadentane systems have been successfully
approached using ACE chemistry on the bis-epoxide 73.
The feasibility of this approach was established using the coupling
of naphthonorbornadiene 5 with bis-epoxide 73
which provided the CO4C-dentane 74
(Scheme 22). The stereochemistry of the coupling protocol was
established by NMR spectroscopy, where the C2v-symmetry combined
with the typical downfield shift of the methylene bridge proton
Ha confirmed its proximity with the oxygen bridge (steric compression)
as required in the assigned structure 74.
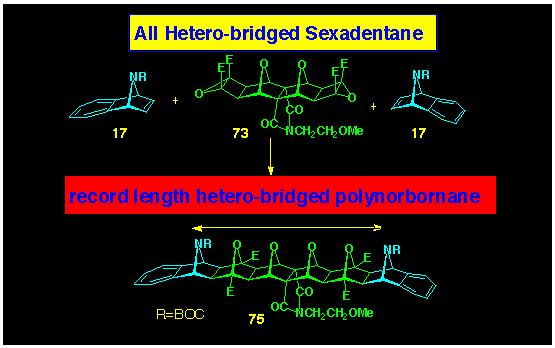
In a related reaction with bis-epoxide 73,
it was found that 7-azabenzonorbornadiene 17 produced a
single 2:1-adduct 75 (Scheme 23). The dynamics of the
N-bridges in this product precluded the use of NMR spectroscopy
to confirm the structure, and its assignment rests on the analogy
provided in the previous reaction with the carbon-bridged dipolarophile.
Notwithstanding, structure 75 is the largest of the hetero
polynorbornanes we have yet prepared and contain six contiguous
7-heteronorbornanes.