In the previous post, I showed how modelling of unbranched alkenes depended on dispersion forces. When these are included, a bent (single-hairpin) form of C58H118 becomes lower in free energy than the fully extended linear form. Here I try to optimise these dispersion forces by adding further folds to see what happens.
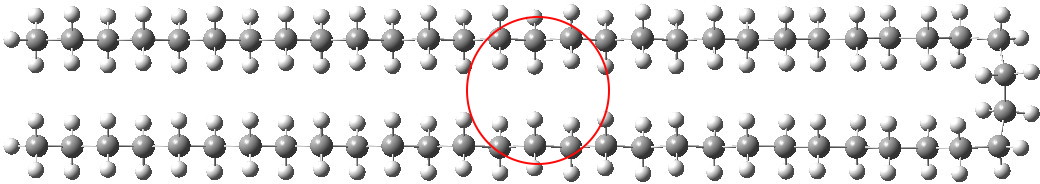
I had noted a small kink in the bent single-hairpin form (above, red circle). What about making a full bend at that point? Such forms have been previously investigated using OPLS-AA mechanics[1], with the finding that such a triple-hairpin conformation (below) was 9.7 kcal/mol higher in energy than the single hairpin (above). OK, its got eight gauche-turns more (four per bend, and which do cost energy), but it also has three rather than just one row of close dispersion-stabilising contacts to compensate. Using quantum rather than molecular mechanics (B3LYP+D3/TZVP), I found that this triple-hairpin folded form was 3.2 kcal/mol higher in free energy than the single hairpin.[2]

Click for 3D
One folded at a slightly different point (below) was in fact higher 4.7 kcal/mol in energy that the single hairpin,[3] indicating that there is an optimum position for the bend.

Click for 3D
I was convinced better folds could be found. So how about this double-hairpin, but in three dimensions to form a prism so that each chain has just as many contacts as the triple-hairpin, but is achieved with two-fewer gauche-turns? Its free energy[4] is 1.6 2.5 kcal/mol lower than the single-hairpin. It did not feature in the previous report[1] and hence represents a new lowest-energy folding (the colour indicates three ribbons of attractive non-covalent interactions, using the NCI technique). I would point out that such “manual” searching for better folds is not really sustainable; a statistical method would normally be used (MD or Monte-Carlo).
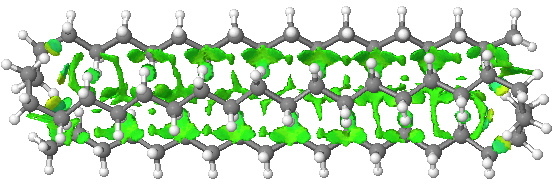
Click for 3D
A similarly folded version of the triple-hairpin can be made (below), with more opportunity for five rows of close dispersion contacts. This time however, the free energy is 1.9 kcal/mol higher than the single hairpin[5] (but the position of the fold does need to be optimised and perhaps a better one can be found). This result does imply that there is an optimum balance between the energy penalty of creating four gauche-turns per fold and the additional energy stabilisation of the dispersion. Perhaps the triple hair-pin above is close to that optimum?
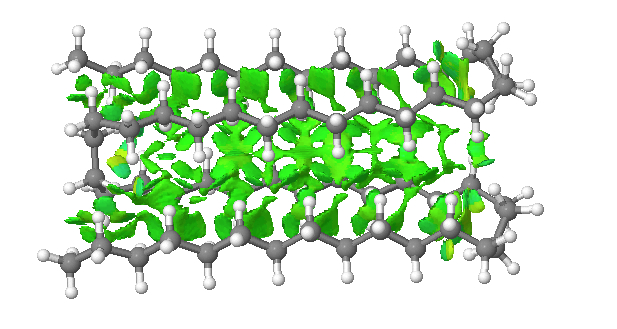
Click for 3D
Unfortunately no crystal structures for the higher linear alkanes have been reported that would give us a reality check on any of these models. Can it really be that difficult to crystallise such molecules?
Author
References
- L.L. Thomas, T.J. Christakis, and W.L. Jorgensen, "Conformation of Alkanes in the Gas Phase and Pure Liquids", The Journal of Physical Chemistry B, vol. 110, pp. 21198-21204, 2006. https://doi.org/10.1021/jp064811m
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.988335
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.988334
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.988771
- H.S. Rzepa, "Gaussian Job Archive for C58H118", 2014. https://doi.org/10.6084/m9.figshare.988333
Tags: energy, energy penalty, energy stabilisation, free energy, lowest-energy folding