Previously on this blog: modelling the reduction of cinnamaldehyde using one molecule of lithal shows easy reduction of the carbonyl but a high barrier at the next stage, the reduction of the double bond. Here is a quantum energetic exploration of what might happen when a second LAH is added to the brew (the usual ωB97XD/6-311+G(d,p)/SCRF=diethyl ether).

In a comment at the end of the first post on this theme, I had noted some crystal structures containing in effect HxAl.Li(OR)y units (x=3,4; y=0-3), noting the variety of structural motifs. The current exploration does not even attempt to cover this range of possibilities, but it is informed by the types of weak interaction that these structures reveal. I will nevertheless accept that whatever pathway is revealed here is likely to represent an energetic upper bound and recognise that lower energy pathways may well exist but are yet to be explored.
{kind=link}
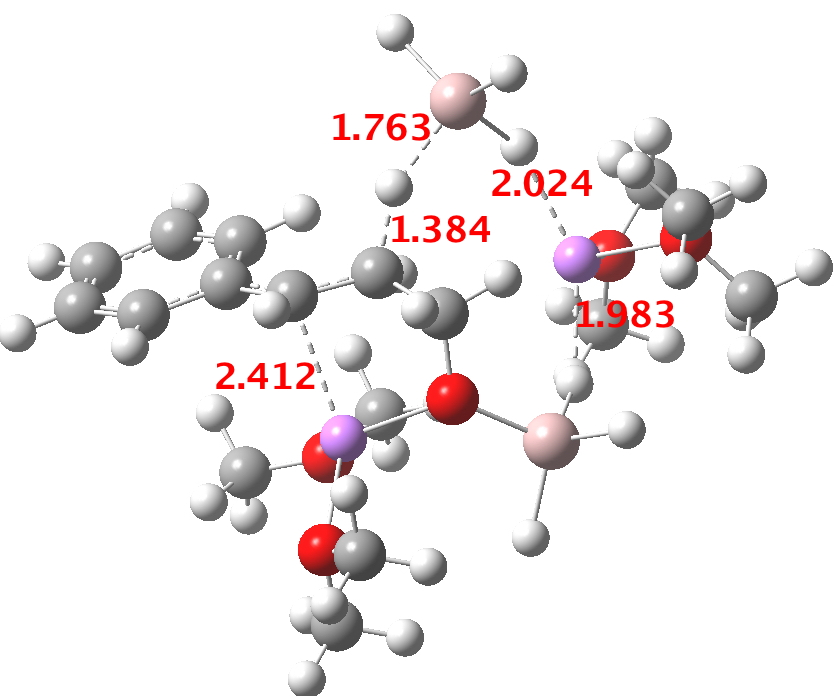
- At the I12 stage, a second AlH4–.Li(OMe)2 is added and hydride transfer occurs antiperiplanar across the C=C bond (TS34-1). The computed free energy barrier ΔG298† is ~24 kcal/mol. The magnitude of this barrier corresponds to a relatively slow reaction occurring around room temperatures or slightly higher.
TS. Click for 3D
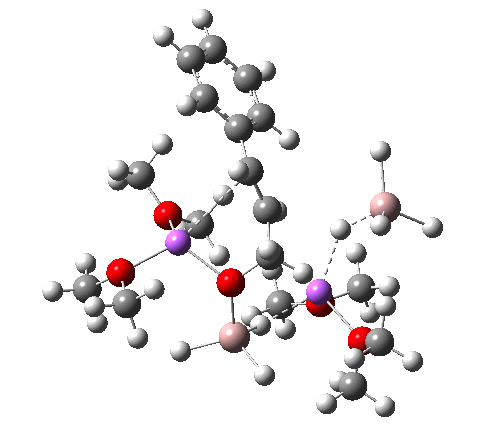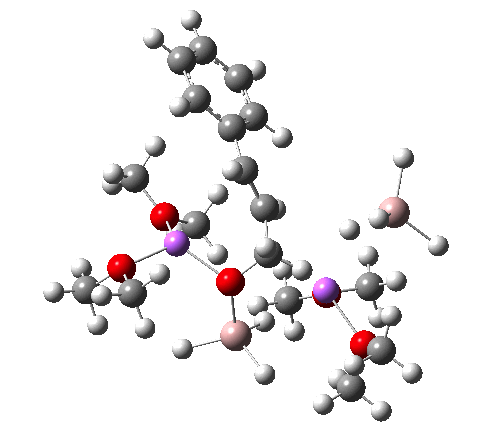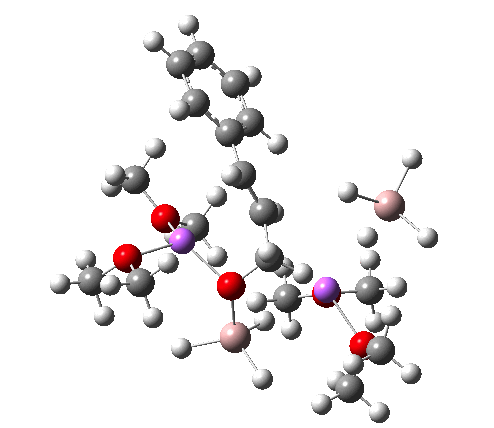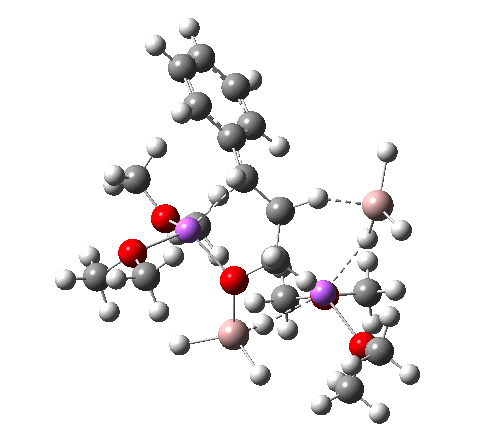
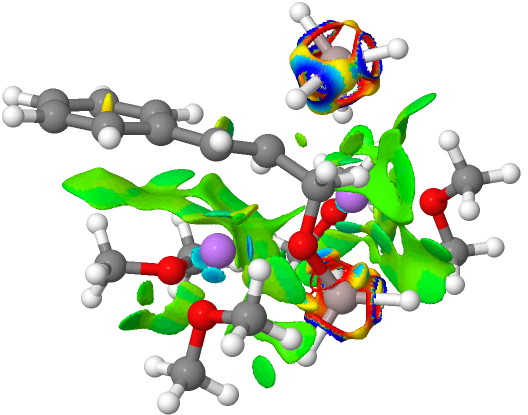
NCI Isosurface (green regions are dispersion stabilizing) Click for 3D
- A transient shallow intermediate I34-1 is formed in which the benzylic anion is stabilised by an adjacent solvated Li centre. The energy of this species (Table below) needs some explanation.‡ Can its free energy really be 1.5 kcal/mol higher than that of the preceding transition state? Yes, because its entropy is lower! The transition state is located on a total energy surface, which does not include thermal and entropic corrections; these are always applied AFTER the stationary points are located. If one inspects these total energies, I34-1 emerges as 1.2 kcal/mol lower than the preceding transition state. This sort of result serves to remind us of the dynamic nature of a potential energy surface, and that static energies may on occasion lead to odd results. Its geometry is shown below, and this too has an interesting feature. The C-H bond just created from the LAH is antiperiplanar to the benzylic anion (locked anti by the Li) and the resulting stereoelectronic effect reduces its C-H calculated[1] stretching wavenumber from the normal value of ~3100 cm-1 to 2231 cm-1, a remarkable reduction.
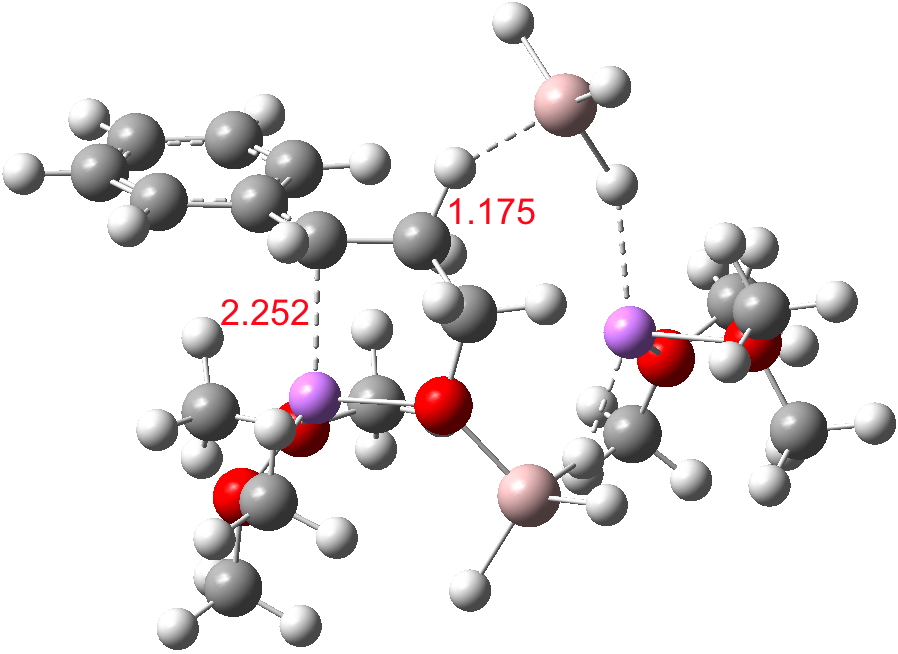
I34-1. Click for 3D
- The C-O-AlH3.Li(OMe)2 ligand now needs to rotate to I34-2 so that metal exchange on the benzylic carbon can occur, with Al displacing Li at that position. As with I34-1, the free energy of this species is actually slightly higher than that of TS34-1. Two AlH3 groups now exist at this stage (each of them formed by hydride donation as part of the reduction process, see below). A hydride transfer metathesis between them (H2Al-H-Al3 is actually a stable bridged species) will generate an AlH2 as part of the 5-ring aluminate ester in P34 and regenerate a molecule of LAH. Transition states for these processes (i.e. TS34-2) proved difficult to locate;† it may be that the ligand rotation and the hydride metathesis are part of the same concerted process but that is not proven yet.
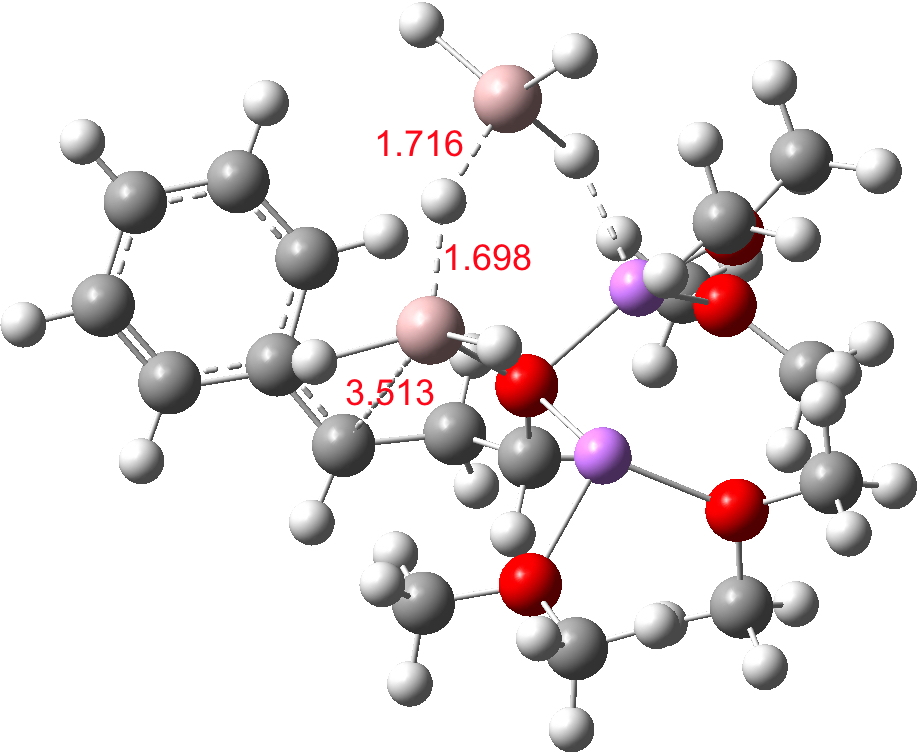
I34-2. Click for 3D
- The final product prior to hydrolysis is appropriately exoenergic.
- I would also remark that many aspects of this reaction remain unexplored. For example, AlH4 can deliver up to four hydrides, becoming progressively substituted as Al(OR)nHy and in the process loosing Al-H…Li weak interactions. What influence this has on the barriers remains unknown.
| Species | Relative ΔG298, kcal/mol | FAIR Data-DOI |
|---|---|---|
| I12 | 0.0 | [2] |
| TS34-1 | 24.1 | [3] |
| I34-1 | 25.5‡ | [1] |
| I34-2 | 25.0‡ | [4] |
| P34 | -8.8 | [5] |
In summary, the first step in the reduction of cinnamaldehyde to cinnamyl alcohol requires just one molecule of “LiAlH4” as reductant and has a very low barrier to reaction. To construct a reasonable model to account for the slower further reduction of the C=C bond requires adding a further LiAlH4, the key feature being the availability of a lithium centre to stabilise out the forming benzylic carbanion. No doubt even better models might include the effects of adding e.g. a third molecule of LAH, and a much more extensive exploration of the various conformational options. But I think the present model might be good enough to augment the apparently relatively limited mechanistic speculations found in text books on the topic.
†You sometimes see this phrase in articles reporting transition state location. What is means it that I tried a half-dozen what I thought were reasonable possibilities, and none of them satisfactorily converged. This semi-random exploration of the potential energy surface revealed a very flat energy potential, with lots of conformational possibilities. At this point, you have to decide whether it is worth the time to continue hunting.
Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
References
- H.S. Rzepa, and H.S. Rzepa, "C 17 H 40 Al 2 Li 2 O 5", 2015. https://doi.org/10.14469/ch/191178
- H.S. Rzepa, and H.S. Rzepa, "C 17 H 40 Al 2 Li 2 O 5", 2015. https://doi.org/10.14469/ch/191172
- H.S. Rzepa, and H.S. Rzepa, "C 17 H 40 Al 2 Li 2 O 5", 2015. https://doi.org/10.14469/ch/191177
- H.S. Rzepa, and H.S. Rzepa, "C 17 H 40 Al 2 Li 2 O 5", 2015. https://doi.org/10.14469/ch/191181
- https://doi.org/
Tags: computed free energy barrier, energy, energy surface, final product, flat energy potential, free energy, lower energy pathways, metal exchange, pence, potential energy surface, reduction, Yes