Following on from my template exploration of the Wilkinson hydrogenation catalyst, I now repeat this for the Grubbs variant of the Alkene metathesis reaction. As with the Wilkinson, here I focus on the stereochemistry of the mechanism as first suggested by Chauvin[1], an aspect lacking in eg the Wikipedia entry. As before, the diagram below is hyperlinked to the appropriate data repository identifier so that you can go straight from the scheme to the data (Top level Data DOI: [2]).

The essence of the reaction is the formation of a metallacyclobutane intermediate, which being approximately symmetrical with a plane of symmetry, can revert to the catalyst and an alkene in one of two ways, reforming the original alkene (two red dot carbons) or a forming a methathesised alkene (red-blue dot carbons).
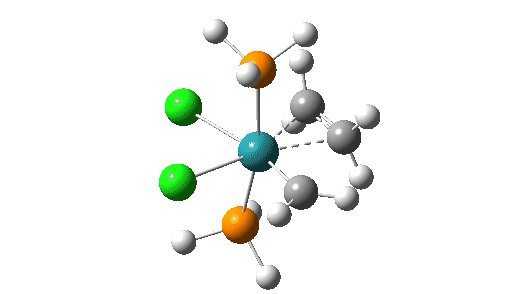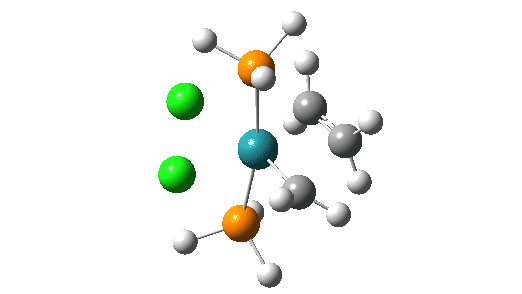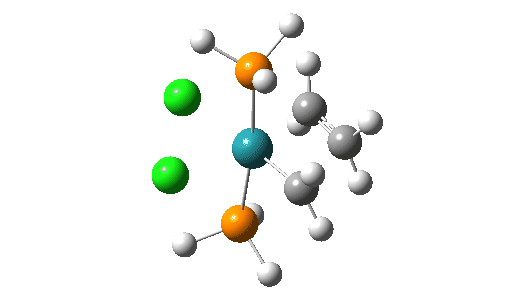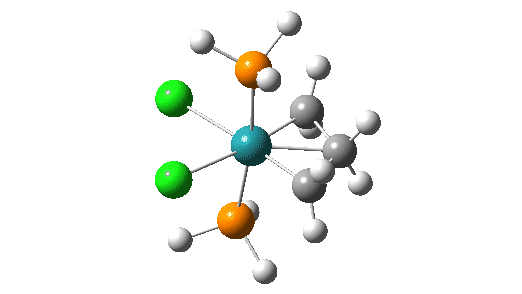
Although the mechanism is often described as a [2+2] cycloaddition in which d-orbital participation from the metal lowers the activation energy significantly, calculations at the MN15L/Def2-TZVPPD/SCRF level indicate there can be up to four discrete steps involved in the process. There are three routes involving these steps that the calculations (B3LYP+GD3+BJ/Def2-TZVPPD/SCRF=DCM) reveal (DataDOI [2]). The starting point for all three routes is the most stable reactant catalyst (left above) which has the H-C-H carbene group in the same plane as the P-Ru-P atoms.
- The red route involves the following steps:
- Activation of the catalyst by rotation of the carbene from its lowest energy orientation by 90°.
- followed by addition of an alkene to form a π-complex –
- then formation of a C-C bond between the alkene and the carbene (animation below). The remarkable feature of this third step is that the carbene group must again rotate through 90° (indicated with a red rotational arrow in scheme above) prior to finally forming the C-C bond.
- The magenta route involves only one step, in which addition of the alkene is directly followed in a second stage by C-C bond formation, via what is called the “hidden intermediate” of the alkene complex (visible at ~IRC -3 for the energy profile below)

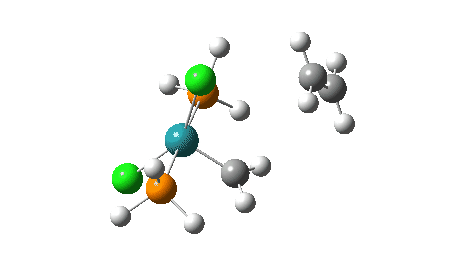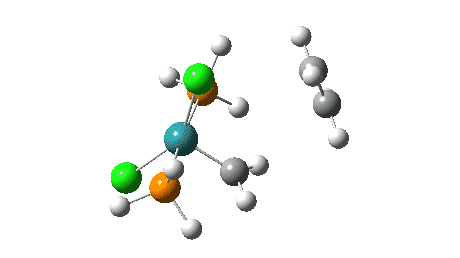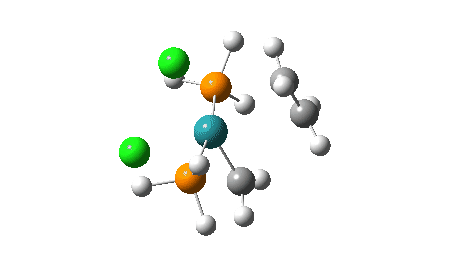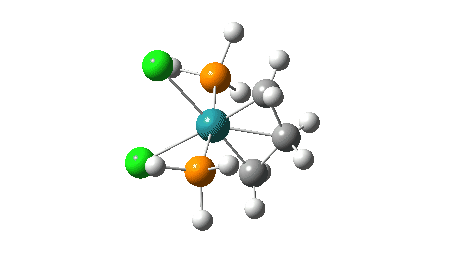
- The green and final route again involves up to four steps:
- A pseudorotation to place the two chlorine atoms di-axial,

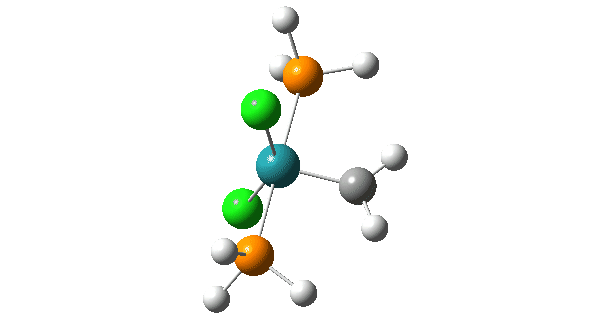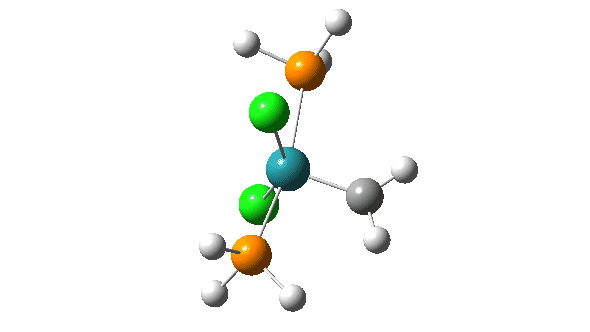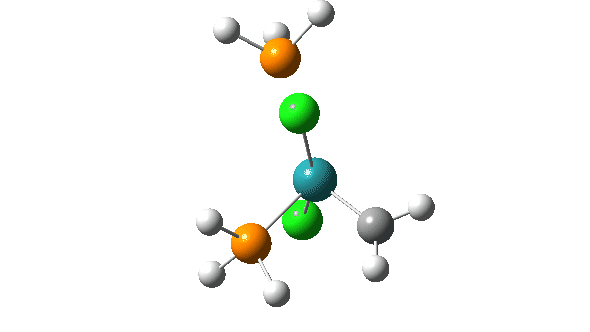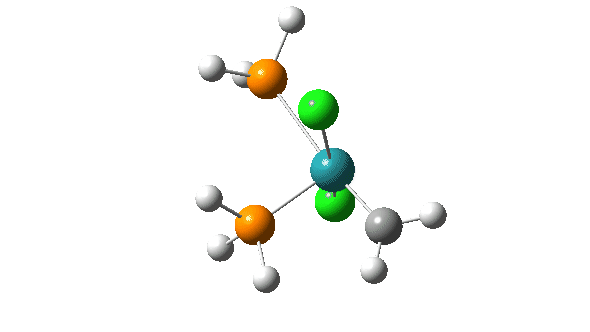
- next, addition of an alkene to form a π-complex
- immediately followed C-C formation between alkene and carbene, again with twisting of the carbene group in the final step. The combined IRC for the last two of these steps (below) shows that the alkene π-complex in fact sits in shallow but real minimum, compared to it being only a “hidden intermediate” in the magenta route.

- The reaction can either reverse as this stage to eliminate a different alkene, or progress through one final pseudorotation step to rejoin the product of the red and magenta routes.
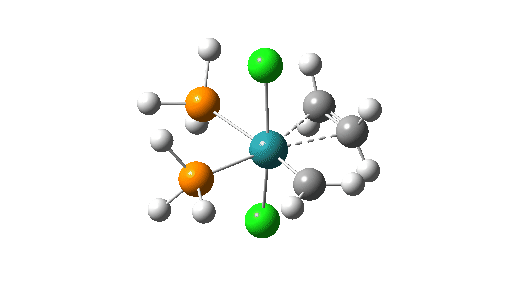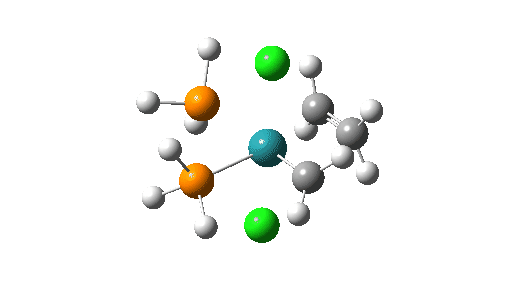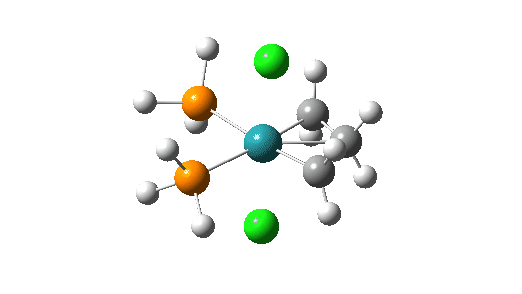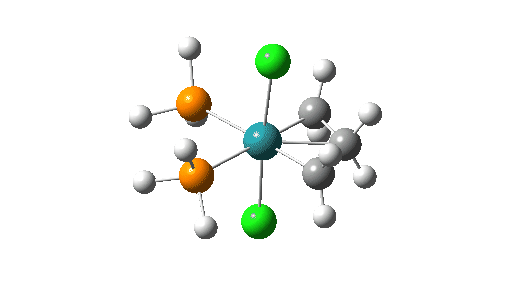
Click on image above to get 3D model of the transition state.
- A pseudorotation to place the two chlorine atoms di-axial,
Of the three routes, the green one has the lowest “high energy” point, corresponding to a barrier of ΔG‡ 14.7 kcal/mol, which corresponds to the facile room temperature reaction it is. The two almost-equal high points are the initial pseudorotation and the alkene complexation, although the final C-C bond formation is also very similar in energy.
So we have learnt that this mechanism is actually a bit more complex than is normally shown and that two of the steps (red and green) involve a very unusual methylene rotation accompanying the C-C bond formation. No doubt, the stereoelectronic orbital interactions responsible for this are fascinating, but an analysis of these will have to wait for another post.
Author
References
- P. Jean‐Louis Hérisson, and Y. Chauvin, "Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d'oléfines acycliques", Die Makromolekulare Chemie, vol. 141, pp. 161-176, 1971. https://doi.org/10.1002/macp.1971.021410112
- H. Rzepa, "Mechanistic templates computed for the Grubbs alkene-metathesis reaction.", 2024. https://doi.org/10.14469/hpc/13796